











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Molecular recognition processes are fundamental for living organisms, for example carbohydrates interact with nucleic acids and proteins, involving a wide range of biological processes [1-5]. Intermolecular CH/π weak interactions commonly participate in the binding between carbohydrates and lectins (proteins that specifically recognize carbohydrates) [6-9]. Crystallographic data have showed that inside carbohydrate recognition domains (CRDs) of lectins, aromatic residues of Trp, Tyr, His and Phe are mediating the recognition of carbohydrates through CH/π interactions [8,10-14].
CH/π interactions have a large contribution of London dispersion forces, which have an impact on the enthalpic term of the free energy [15,16]. Cooperativity of CH/π interactions is a property that becomes relevant when for example in D-galactose the C-H bonds in positions 3, 4 and 5 interact with the aromatic side chains of proteins [17-20]. The experimentally estimated energy for CH/π interaction for the carbohydrate-aromatic stacking in water is approximately 1.5 kcal mol-1 [(21,22].
Previous studies of the CH/π interactions have been based on the use of diverse analytical, biochemical and biophysical techniques [23-27]. An understanding of all the factors that control this interaction is relevant for the design of improved artificial carbohydrate receptors [28]. CH/π interactions together with hydrogen bond capabilities would result in new highly selective artificial receptors, with potential applications in medicine and materials science [29-36].
In a previous work, we showed that methyl 2,3,4,6-tetra-O-methyl-a-D-galactopyranoside (aMeGal) was able to recognize benzene more efficiently than methyl 2,3,4,6-tetra-O-methyl-a-D-mannopyranoside (aMeMan) [20]. It was found that the value of enthalpy of solvation in benzene for aMeGal was 2.5 kcal mol-1 lower than the enthalpy of solvation of aMeMan. Characterization of the molecular region where the interaction takes place was done by nOe 1H-NMR spectroscopy and it was found that C-H protons H3, H4 and H5 of galactopyranoside were in close contact with aromatic benzene ring. Indicating that benzene was forming an ordered solvation sphere around the carbohydrate.
In this work, we have evaluated the role of CH/π interactions in the molecular recognition of acetyl 2,3,4,6-tetra-O-methyl-β-D-galactopyranoside with 6-substituted 2-methoxypyridine and 2(1H)-pyridones.1H-NMR titrations and chemical shift perturbation experiments were performed to demonstrate the presence of CH/π interactions. Additionally, DFT calculations were performed to support the presence of CH/π interactions and to visualize the possible geometrical arrangement of the formed supramolecular complexes.
Synthesis
The acetyl 2,3,4,6-tetra-O-methyl-β-D-galactopyranoside 4b was synthesized by permethylation of methyl a-D-galactopyranoside 1 using 50% (w/w) sodium hydroxide solution and iodomethane in DMSO. Permethylation was done to avoid undesirable dominant hydrogen bond formation during titration experiments and to make the acetyl monosaccharide 4b less hydrophilic. The permethylated product 2 was obtained in 56% yield and subsequently hydrolyzed using an aq 2.0 N HCl solution to produce a mixture of a and b epimers of monosaccharide 3, in a 3:1 ratio. The last step was the acetylation of 3 in acetic anhydride with sodium acetate at 70 ºC. A mixture of the a and b epimers of acetyl 2,3,4,6-tetra-O-methyl-D-galactopyranoside 4a and 4b, respectively was obtained in 60 % yield, in a 1:3 disatereomeric ratio (Scheme 1). The epimers were separated using silica gel flash chromatography. Carbohydrates products were characterized through 1D and 2D NMR experiments. Full assignment of all proton signals was achieved.
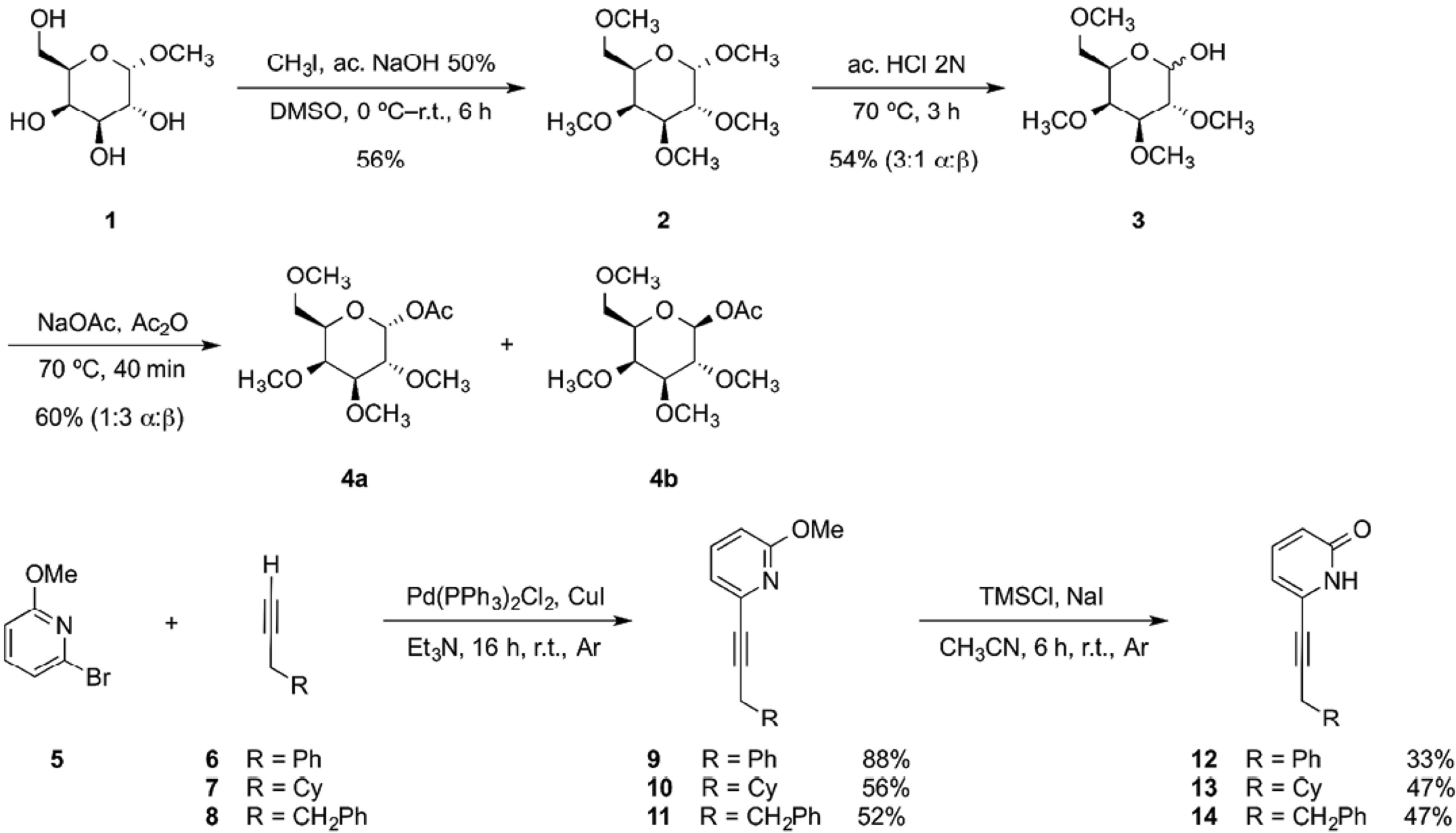
Scheme 1 Synthesis of acetyl galactopyranoside 4b, 2-methoxypyridines 9-11 and 2(1H)-pyridones 12-14.
The 6-substituted 2(1H)-pyridones were synthesized starting with a Sonogashira coupling [37] between 6-bromo-2-methoxypyridine 5 and the corresponding terminal alkyne (6-8), obtaining 2-methoxypyridines (9-11) in good yields. The 6-substituted 2(1H)-pyridone receptors (12-14) were obtained in modest yields by adding TMSCl and dry NaI at room temperature in dry acetonitrile (Scheme 1) [38,39].
Crystal structures of 2(1H)-pyridones (12-14).
The crystal structures of 2(1H)-pyridones (12-14) were obtained to confirm their molecular structure (Fig. 1).
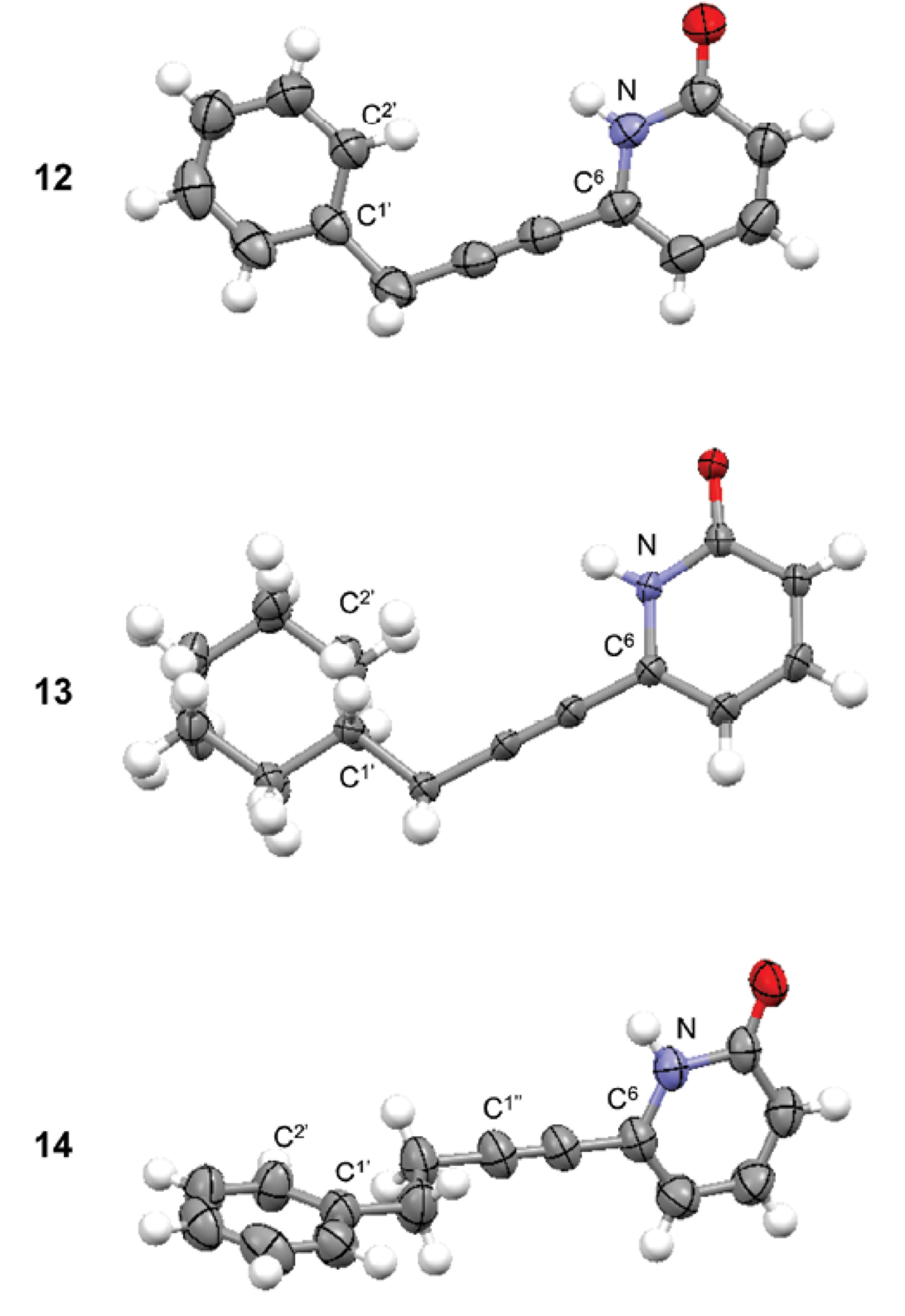
Receptor 12 had a planar structure (dihedral angle N-C6-CH2-C1’ q = 2.4º); phenyl and 2(1H)-pyridone groups were practically coplanar (dihedral angle N-C6-C1’-C2’ q = -5.9º). Phenyl group was eclipsed with N-C6 bond. Receptor 13 had very similar conformation than receptor 12. Cyclohexyl and 2(1H)-pyridone groups were coplanar (dihedral angle N-C6-C1’-C2’ q = -17.0º) with cyclohexyl group in eclipsed conformation with N-C6 bond. In receptor 14, the phenyl group was oriented perpendicular with respect to the 2(1H)-pyridone group (dihedral angle N-C6-C1’-C2’ q = -90.5º). Additionally the conformation of substituents along -CH2-CH2- segment was anti (dihedral angle C1’-CH2-CH2-C1’’ q = 174.7º). The conformation of the receptor will be important for the carbohydrate recognition.
1H-NMR Chemical Shift Perturbation Data Analysis
The 1H-NMR upfield shift of specific ring protons in monosaccharides upon addition of an aromatic compound, such as benzene or toluene, has served as a direct evidence for the presence of CH/π interactions in molecular recognition [20,40,41]. In galactose exists a hydrophobic patch on the alpha face, where it is possible for the electronic density of p orbitals of the aromatic ring to interact through CH/π interactions [21]. The 2-methoxypyridne receptors 9-11, were used to demonstrate the existence of CH/π interactions in the molecular recognition of sugar 4b. The effect of the addition of 32 equivalents of 2-methoxypyridine receptors on the 1H-NMR chemical shift variation for the ring protons of acetyl galactopyranoside 4b is shown in Table 1.
Table 1 Shielding (Hz) measured for the resonance signals of the ring protons of acetyl galactopyranoside 4b upon addition of 32-fold excess of 2-methoxypyridine receptors (9-11).[a],[b],[c],[d],[e]
[a] Shielding determined at 750 MHz and 298 K in CDCl3. [b] TMS signal was used as internal standard. [c] The concentration of galactopyranoside 4b was 0.01 M. [d] Shielding was calculated with formula ∆δgal = δ - δinitial. [e] Each value is the average of three different determinations. A minus sign indicates up-field shift; a plus sign indicates down-field shift.
The anisotropy of benzene ring would lead to up-field shift of proton signals of the galactopyranoside 4b [( 40]. The extent of such up-field shift would be correlated to the closeness of the π electrons with the protons of 4b. The ring proton signals H3, H4 and H5 of substrate 4b were up-field shifted upon addition of receptors 9 and 11. For both receptors proton signal H4 resulted the most up-field shifted, with values of -7.0 and -6.3 Hz for receptors 9 and 11, respectively. These values were close to those reported by Fernández-Alonso et al. [41], where they reported an up-field chemical shift for signal H4 of methyl b-D-galactopyranoside of -6.3 Hz when adding a 0.2 M phenol solution to a 0.01 M solution of the sugar in D2O. It is important to mention that this effect would not be a result of the inherent perturbations of the change in concentration of the solution since proton signal H2 was down-field shifted; thus implicating the existence of specificity in the solvation process.
The molecular recognition of 4b was orchestrated by CH/π interactions taking place at the hydrophobic patch of the alpha face, with protons H3, H4 and H5 and the phenyl group of receptors 9 and 11. When receptor 10 was added to substrate 4b, the up-filed shift values for protons H3, H4 and H5 were diminished, since the replacement of phenyl group with cyclohexyl group (in the molecular structure of receptor 10) would eliminate CH/π interactions. The solvation effect of these receptors towards galactopyranoside 4b would most likely take place in an ordered fashion. The 1H-NMR spectra of 4b before and after the addition of receptor 9 are depicted in Fig. 2 (for the addition of receptors 10 and 11 see Figures S1 and S2, respectively in supporting information).
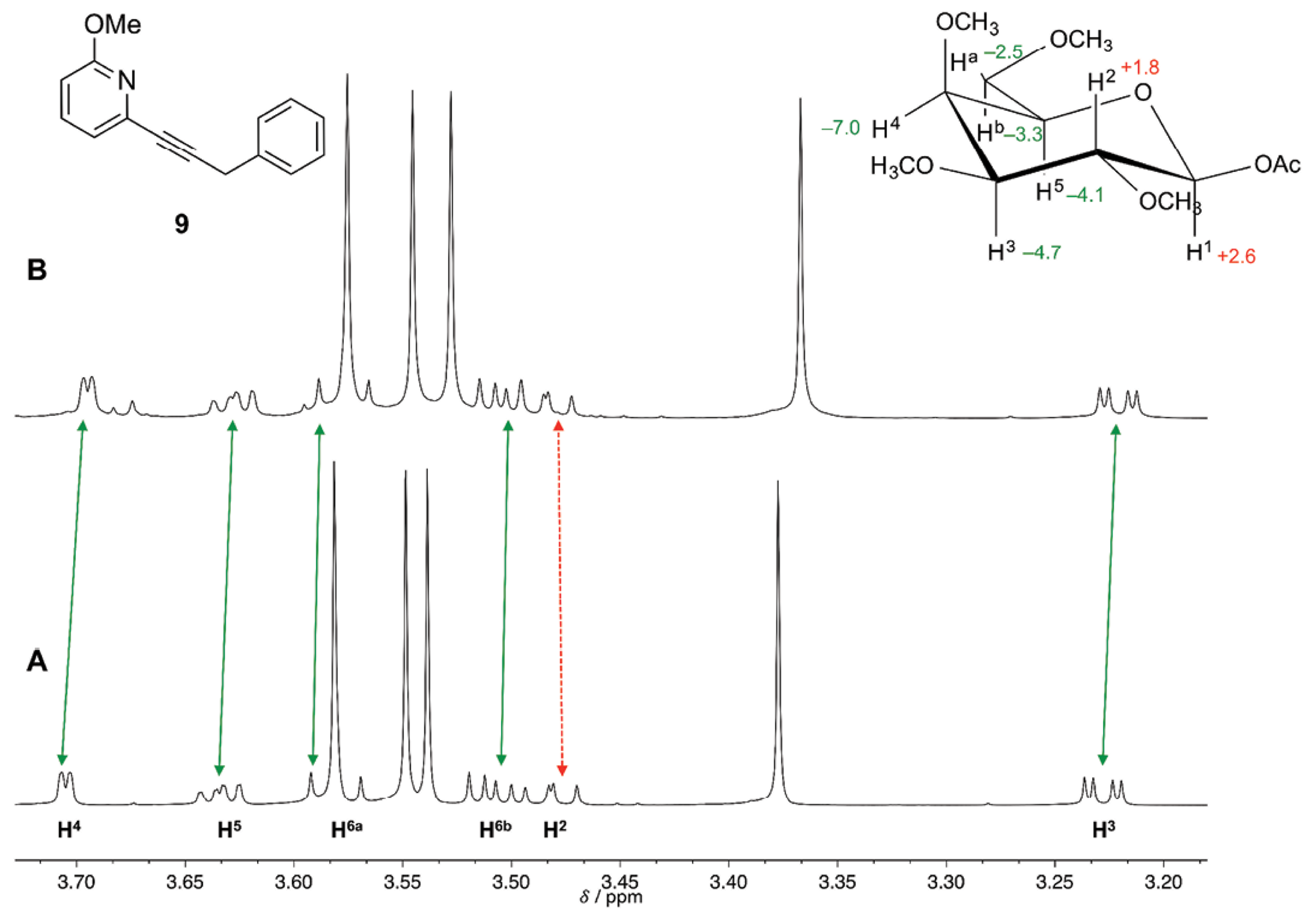
Fig. 2 A) 750 MHz 1H-NMR spectrum of galactopyranoside 4b 0.01 M in CDCl3 (volume 0.5 mL) at 298 K, and B) upon addition of 32-fold excess of receptor 9. The value of the change in chemical shift (in Hz) for the protons of substrate 4b is indicated in the inset. A minus sign indicates up-field shift (indicated by continuous arrows) and a plus sign indicates down-field shift (indicated by dashed arrow). TMS (0.0 ppm) was used as internal standard.
Affinity Constants Determination
The 1H-NMR titration curves for 2-methoxypyridine receptors 9-11 and substrate 4b for the determination of affinity constants resulted in straight lines, meaning that association was very poor. Therefore with 1H-NMR it was not possible to determine the K a values for those receptors. For the association between substrate 4b and 2(1H)-pyridone receptors, 1H-NMR titrations were performed in order to assess the strength of the molecular association mediated by weak CH/π interactions. The galactopyranoside 4b was used as the titrant, while the concentration of 2(1H)-pyridone was kept constant during all the titration. 2(1H)-pyridones were not added to 4b because it is known that 2(1H)-pyridones have the ability to form supramolecular aggregates (such as dimers, trimmers, etc.) in solid state and in solution [42-47]. It was necessary for the titrations to add a large amount of equivalents of galactopyranoside (up to 70-fold excess) in order to obtain a curve that could be properly adjusted (and to reach a plateau in the curve). This methodology was followed as the one reported by Anthony Daivs et al. where initial receptor concentration was set to 0.25 M and added 1066-fold excess equivalents for sensing D-galactose [48]. The obtained curves (∆δ vs [4b]) were adjusted to non-linear fitting for 1:1 stoichiometry complex formation with global analysis. The curve obtained for receptor 12 is shown in Figure 3, the signals shown are the ones used for the global analysis [49]. The good adjustment of the curves showed that there was no other association processes than the 1:1. The obtained K a values are presented in Table 2.
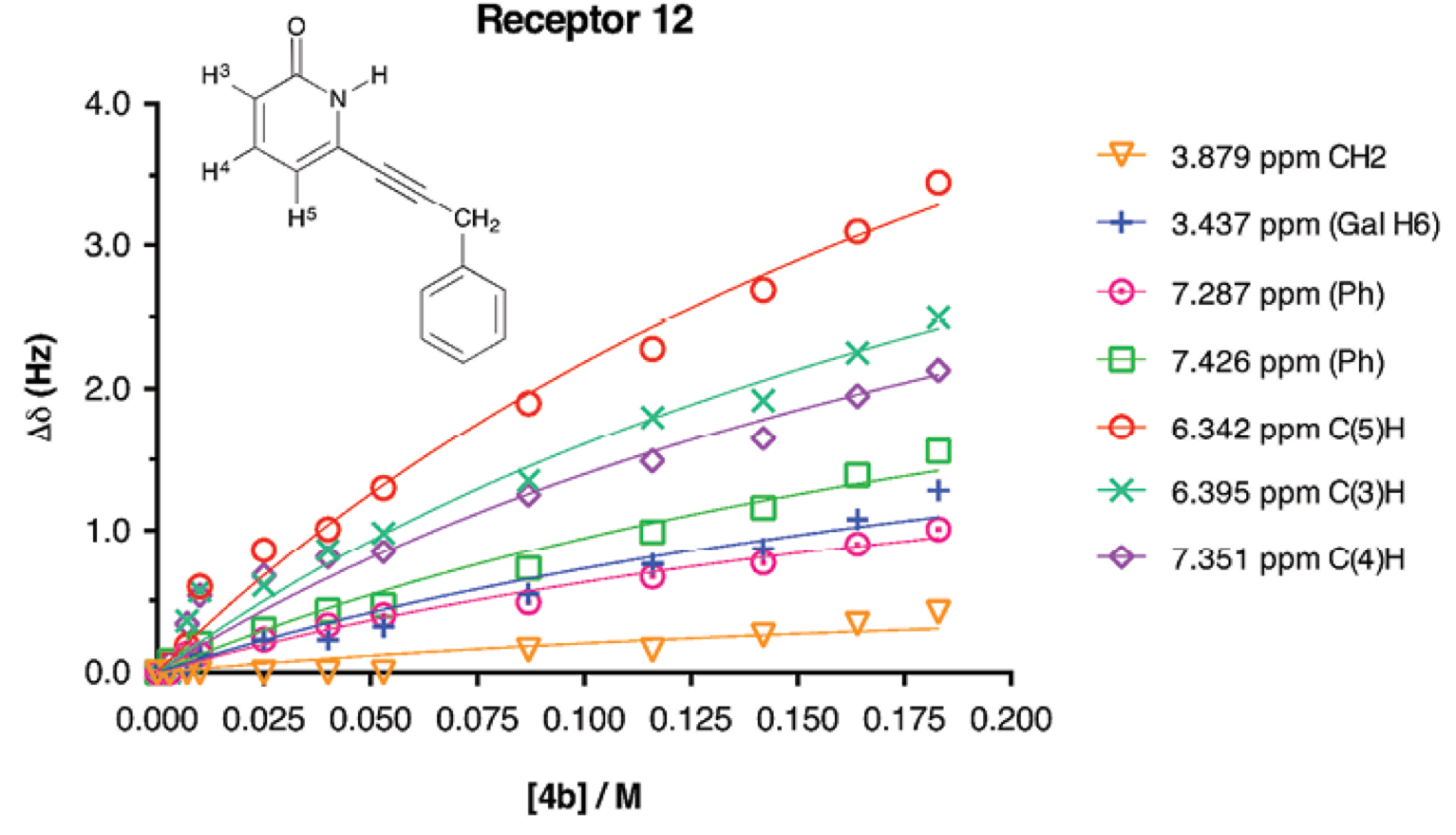
Fig. 3 Experimental values for the NMR binding study of receptor 12 + galactopyranoside 4b in CD3CN. [12]initial = 10 mM, [4b]titrant = 183 mM. The non-linear adjustment is shown.
Table 2 Affinity constant values K a determined for the formation of galactopyranoside-2(1H)-pyridone supramolecular complexes.[a],[b],[c]
[a] Data were obtained from 1H-NMR 750 MHz at 298 K in CD3CN. [b] TMS signal was used as internal standard. [c] Galactopyranoside 4b aliquots were added to a 0.01 M 2(1H)-pyridone solution. The concentration of galactopyranoside 4b was 0.01 M. Proton signals of 2(1H)-pyridone were evaluated in global analysis adjustment.
Although the K a values resulted low, it was possible to observe the effect of the CH/π interactions in the association. Receptors that had a phenyl group (receptors 12 and 14) had higher K a values than receptor that contained cyclohexyl group (receptor 13). This difference in K a shows the ability of receptors 12 and 14 to form CH/π interactions with the galactopyranoside substrate 4b. In a very important contribution, Jiménez-Barbero et al. have reported low affinity values K a for aromatic-carbohydrate CH/π interactions and has mentioned that weak affinity values that should be used as qualitative [40].
Computational Studies
We performed theoretical calculations to evaluate the recognition process and to define the structure of the molecules to be synthesized [27]. Calculations were done using Gaussian 09 software [50]. The acetyl b-D-galactopyranoside−2(1H)-pyridone supramolecular complexes were optimized at the M06-2X/6-31+G(d,p) level, since the M06-2X hybrid meta-generalized exchange-correlation functional is specially designed to account for dispersive interactions [51-54]. The 6-31+G(d,p) basis set was used because addition of diffuse functions to double split valence basis has shown to be more important than increasing to a triplet split valence basis when calculating reaction energies and activation energies with DFT [55]. In order to eliminate the BSSE (basis set superposition error) during the optimization the counterpoise command was used [56]. These calculations were performed on a solvent free model and acetyl b-D-galactopyranoside was optimized without permethylation of hydroxyl groups [20].
The molecular graphic of 12-4b supramolecular complex is shown in Fig. 4. On one side of the complex, it was observed that 2(1H)-pyridone and the acetyl groups were in close proximity. In such manner, hydrogen bond interactions such as C=O---HC (2.35 Å) and NH---OC (2.24 Å) could be formed. On the other side of the complex the closest distance between C3-H bond of 4b and the phenyl group was 2.38 Å with a C3H---Ph angle of 163.6º, representing the possible CH/π interaction.
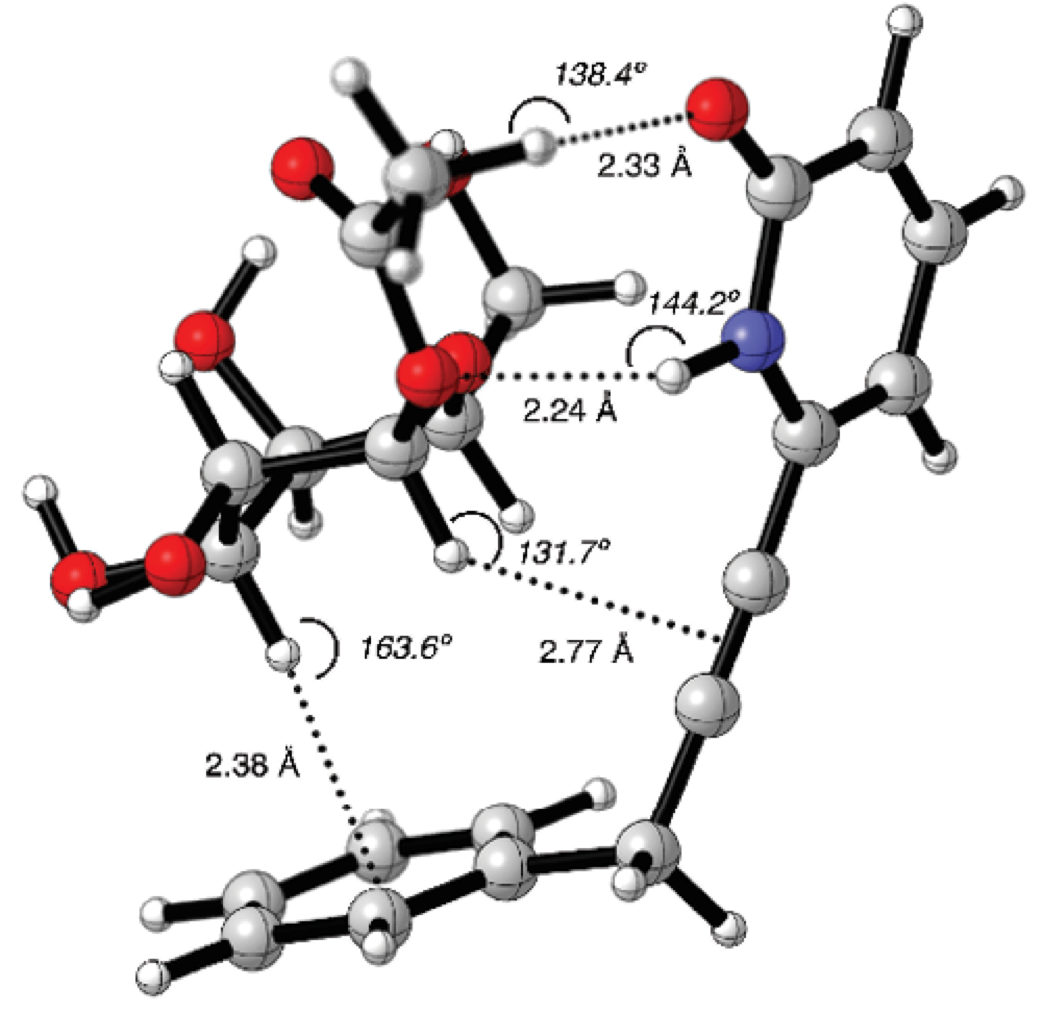
Fig. 4 Counterpoise corrected geometry optimization for the supramolecular complex 12-4b. Key intermolecular distances are given in Å and angles are given in º.
Another possible CH/π interaction could be formed between C1-H bond and -C≡C- group, with a distance of 2.77 Å and an angle of 131.7º. The dihedral angle N-C6-CH2-C1’ was q = 87.3º, the receptor 12 formed a cavity where the carbohydrate 4b could place the alpha face C-H bonds pointing towards the aromatic moieties.
The molecular graphic for complex 13-4b is shown in Figure 5. The cyclohexyl group has no possibility to form CH/π interactions. Interestingly the only possible CH/π interaction that could be observed was between the C1-H and the -C≡C- spacer group, with a distance of 2.79 Å with an angle of 143.8º.
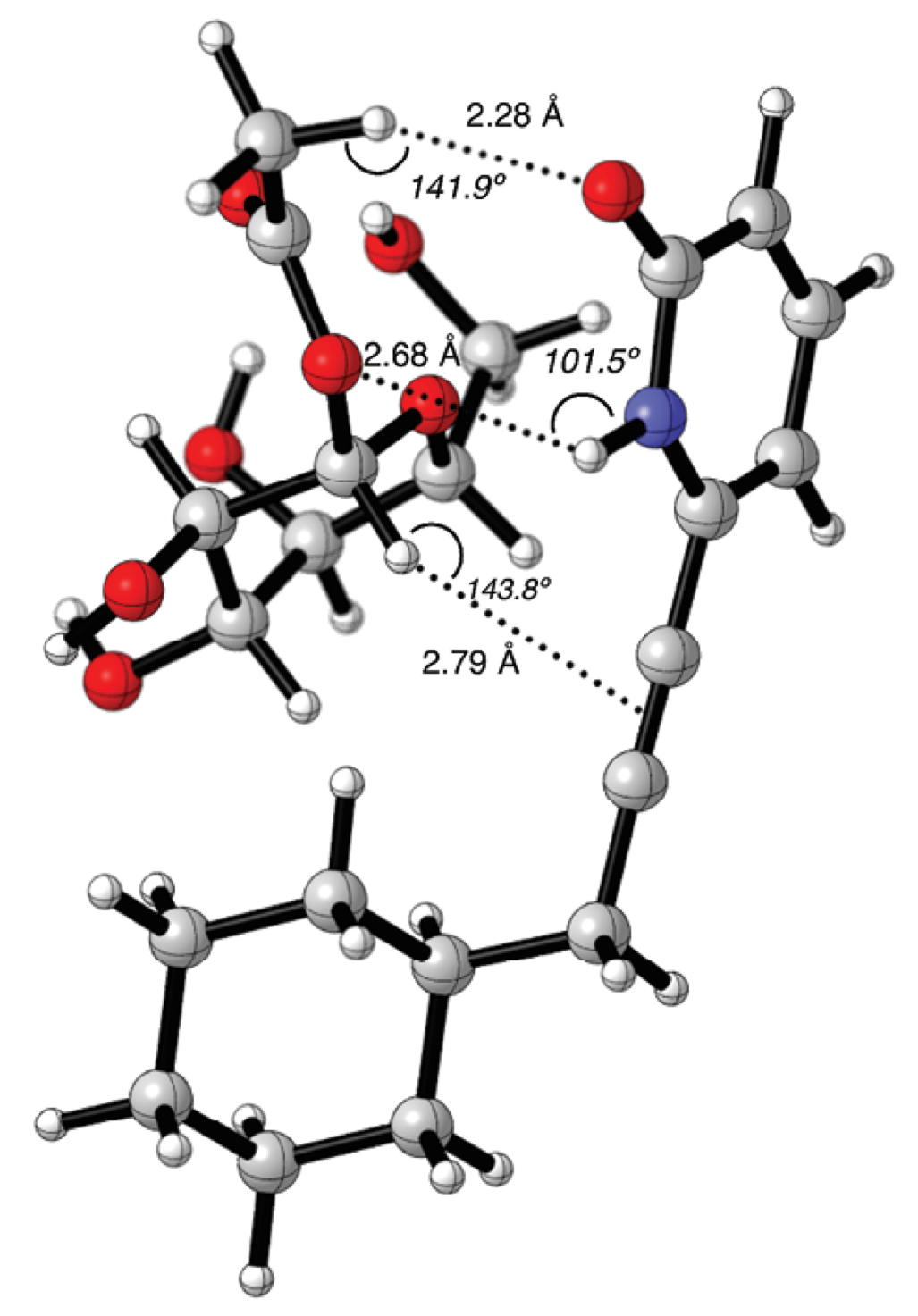
Fig. 5 Counterpoise corrected geometry optimization for the supramolecular complex 13-4b. Key intermolecular distances are given in Å and angles are given in º.
In the molecular graphic of 14-4b supramolecular complex, the carbohydrate’s alpha face was pointing towards the phenyl ring (Fig. 6). The C1’-CH2-CH2-C1’’ segment had an angle of q = 66.8º in gauche conformation. With this angle, it was possible to form a CH/π interaction between the phenyl group and the galactopyranoside C4-H bond, with a distance of 2.86 Å and C4H---Ph angle of 132.1º. The CH/π distance resulted alike to those identified for the fucose-benzene interaction calculated with the MP2 method [41]. The C1-H and C6-H bonds are in close proximity with the -C≡C- group of the pyridone receptor, with distances of 2.83 Å and 2.90 Å, respectively. It would be possible to have more than one type of CH/π interactions in this supramolecular complex [57].
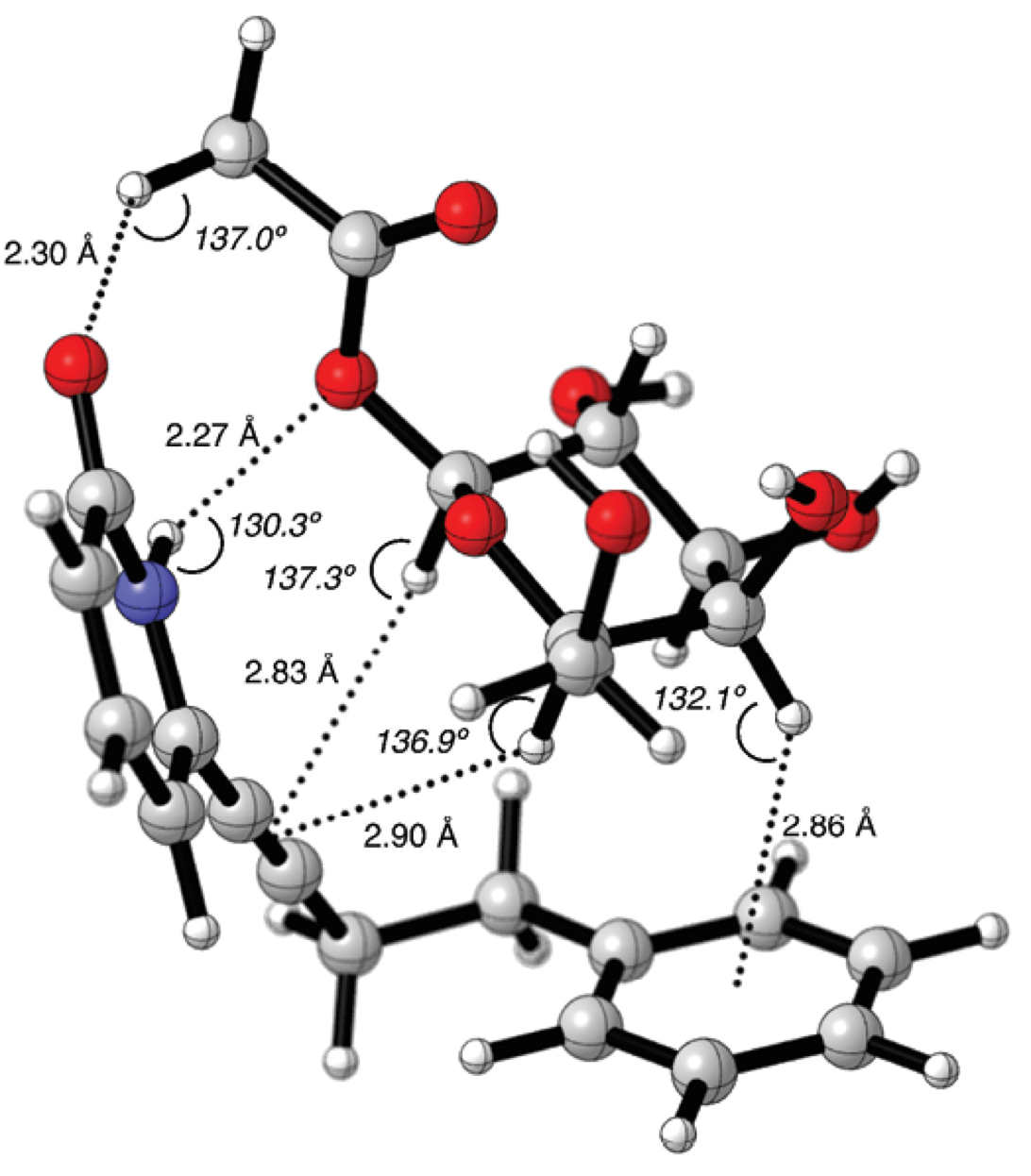
Fig. 6 Counterpoise corrected geometry optimization for the supramolecular complex 14-4b. Key intermolecular distances are given in Å and angles are given in º.
The calculated interaction energies DE of the supramolecular complexes are shown in Table 3. The energy for the supramolecular complex 2(1H)-pyridone-4b is included as reference value (see Fig. S12 in supporting information). Such energy value accounts for the contribution of the hydrogen bond interactions without any CH/π interaction. Supramolecular complexes 12-4b and 14-4b were very similar in energy; they were -3.2 kcal mol-1 more stable than the reference complex 2(1H)-pyridone-4b. This value was in good agreement with the experimentally determined CH/π energy for galactose-benzene interaction [20,26,41]. Whereas 13-4b complex resulted only 0.6 kcal mol-1 more stable than reference complex. Computational optimizations supported the experimental results obtained by 1H-NMR titrations; CH/π weak interactions take place in the molecular recognition of acetyl galactopyranoside 4b.
Table 3 Calculated formation energies for the supramolecular complexes galactopyranoside-2(1H)-pyridone at M06-2X/6-31+G(d,p).[a]
[a] Energies calculated with the BSSE correction. No imaginary frequencies were found.
The CH/π interactions were further demonstrated using software AIMALL [58] with the M06-2X/6-31+G(d,p) theory level. In the Atoms in Molecules theory the criteria for two atoms to interact in a molecule are the presence of a bond critical point (BCP), a space where the electron density is accumulated between two nuclei (density attractors) and a bond path that connects them. In Fig. 7, it is shown the electron density topology for the 14-4b supramolecular complex, where the important CH/π and hydrogen bond BCPs are highlighted.
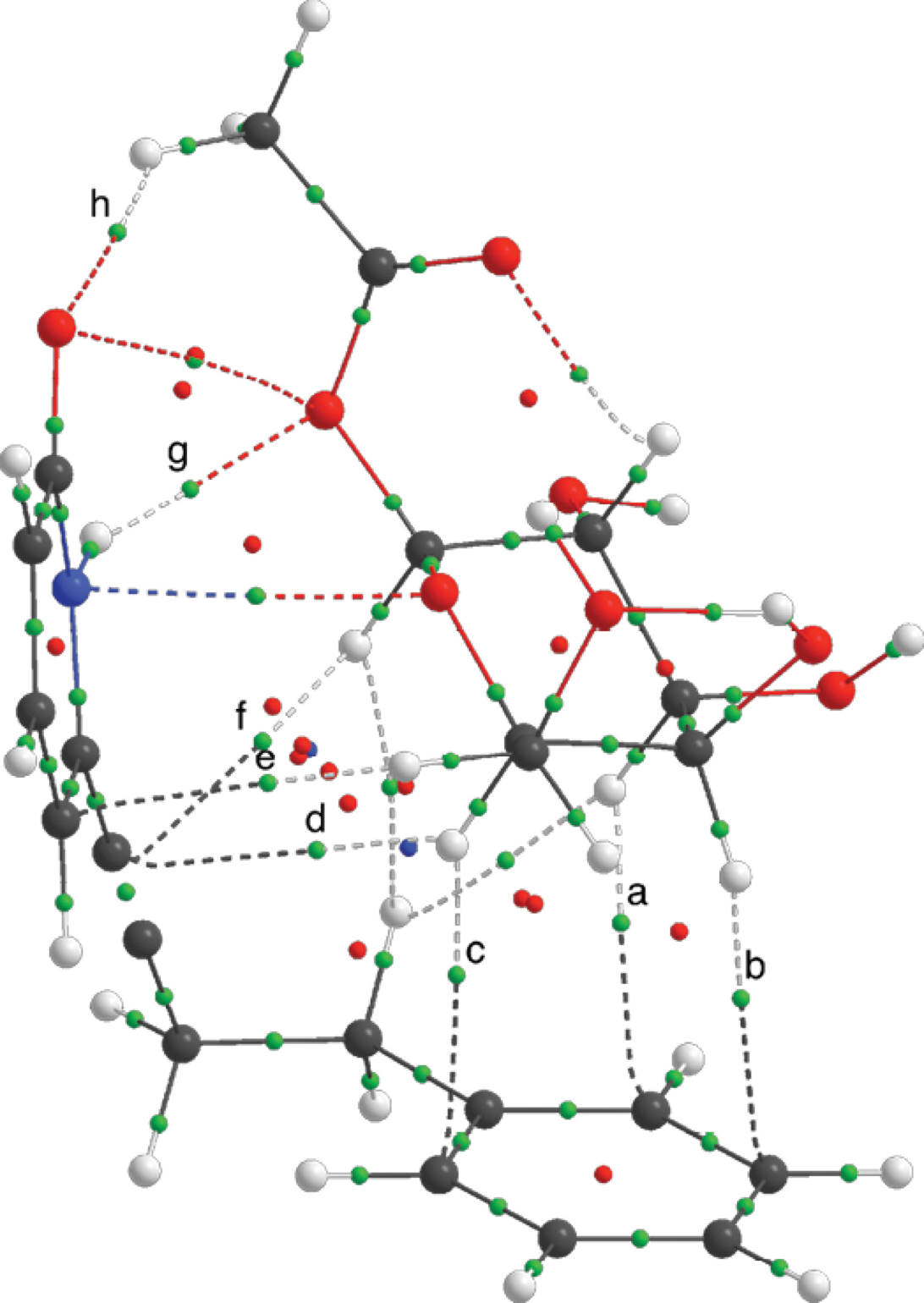
Fig. 7 Electron density topology for the supramolecular complex 14-4b at M06-2X/6-31+G(d,p). Bond critical points for the intermolecular hydrogen bonds and CH/π interactions and marked with letters.
The value of the Laplacian (∇2r) for these BCPs is larger than zero, the electronic charge is delocalized and it belongs to a close-shell interaction. The ellipticity (ε) of the BCP is an indicative on the asymmetric charge distribution in the bond path; non-covalent interactions are characterized to have larger values of ellipticity than covalent stronger interactions (see Table S7 in supporting information). All of the above indicates that the weak interactions that were present in the supramolcular complexes were of non-covalent nature [59]. The values of BCPs for the CH/π for the other complexes had the same trend.
Conclusions
We have studied the participation of weak CH/π carbohydrate-aromatic interactions in the molecular recognition of acetyl 2,3,4,6-tetra-O-methyl-β-D-galactopyranoside 4b by 6-substituted 2(1H)-pyridone receptors. CH/π interactions were demonstrated through 1H-NMR titrations. Key upfield shifts of ring protons H3, H4 and H5 signals in the alpha face of galactopyranoside were observed due to the proximity with 2-methoxypyridine 9 and 11 receptors. No affinity constant values (K a ) could be determined for 2-methoxypyiridine receptors. In the case of 2(1H)-pyridone receptors a plateau value was reached during the 1H-NMR titration and K a values were determined. However, since the association was weak, as expected, these values had to be taken as qualitative. Receptors 12 and 14 showed better affinity to galactopyanoside than receptor 13, since a phenyl ring is present in their molecular structure, enabling CH/π interactions. X-ray diffraction data demonstrated that the conformation of 2(1H)-pyridones in solid state was planar. DFT computational studies were important to understand the association process of the receptors with the galactopyranoside 4b substrate. Conformational changes were necessary in order to form a cavity, where carbohydrate substrate could be recognized. The calculated energy difference that accounted for the CH/π stabilization energy was -3.2 kcal mol-1. This value was in accordance with the one reported for aromatic-galactose interaction. This work has represented a contribution in the field of the development of new receptors for monosaccharides.
Materials and methods
All reagents were used without further purification. Starting materials 6-bromo-2-methoxypyridine 5, 3-phenyl-1-propyne 6, 3-cyclohexyl-1-propyne 7 and 4-phenyl-1-butyne 8 were distilled before use. All solvents used were reagent grade and were dried and distilled following standard procedures. Flash chromatographic purification was performed using silica gel (particle size 0.040-0.063 mm) packed in glass columns; the eluting solvent was determined by thin-layer chromatography (TLC). Melting points were determined on a melting point apparatus. NMR spectra were recorded with 400, 500 and 750 MHz instruments. 1H-NMR chemical shifts are reported in parts per million (ppm) relative to TMS signal (0.0 ppm). Multiplicities are given as: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), ddd (doublet of doublet of doublets), dt (doublet of triplets), qd (quartet of doublets), qt (quartet of triplets), tdp (triplet of double of pentuplets), dddt (doublet of doublet of doublet of triplets), dddq (doublet of doublet of doublet of quartets), m (multiplet), and the coupling constants, J, are given in Hz. 13C-NMR chemical shifts are reported relative to the solvent residual peak (CDCl3, 77.16 ppm). IR frequencies are given in cm-1. Optical rotation [a]D values are given in 10-1 deg cm2 g-1. Mass Spectrometry data were acquired on mass spectrometer adapted with a DART, electronic impact and FAB ion sources. HRMS was performed using an ESI+ ion source. X-ray diffraction studies were realized on a Bruker AXS diffractometer with an area detector, Mo Ka radiation, l = 0.71078 Å. CCDC-1510326 (receptor 12), CCDC-1510325 (receptor 13) and CCDC-1510331 (receptor 14) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.ak/structures. Figures 4, 5 and 7 were made using CYLview visualization software [60].
1H-NMR chemical shift perturbation experiments
A galactopyranoside 4b stock solution (0.01 M) was prepared in CDCl3. A 2-methoxypyridine receptor solution (1.0 M) was prepared using the galactopyranoside 4b stock solution. A volume of the solution of 2-methoxypiridine (32 equivalents) was added to the galactopyranoside 4b stock solution (0.5 mL) contained in a NMR tube. The mixture was well shaken after the addition, and the 1H-NMR spectrum was recorded. Probe temperature was adjusted to 298 K.
General procedure for affinity constants determination
A 2(1H)-pyridine receptor stock solution (0.01 M) was prepared in CD3CN. Solutions (0.1, 0.2, 0.5, 1.0, 2.0 and 5.0 M) of the galactopyranoside 4b were prepared using the 2(1H)-pyridone receptor stock solution. Aliquots of the galactopyranoside solutions were added to the 2(1H)-pyridone stock solution (0.5 mL) contained in an NMR tube. The mixture inside the tube was well shaken after each addition, and the 1H-NMR spectrum was recorded. Probe temperature was adjusted to 298 K. Plots of ∆δreceptor vs. [4b] were obtained. In all cases a 1:1 stoichiometry was obtained for the non-linear data fitting. Each experiment was done in triplicate.
Synthesis of carbohydrates 2-4
Methyl 2,3,4,6-tetra-O-methyl-a-D-galactopyranosoide (2). In a round bottom flask methyl a-D-galactopyranoside 1 (3.0 g, 15.5 mmol) was dissolved in DMSO (20 mL). The mixture was cooled to 0 °C in ice bath and a 50% w/w NaOH aq solution (10 mL, 250 mmol) was added slowly. After a gel suspension was formed, CH3I (27.36 g, 193.0 mmol) was added dropwise and left stirring for 6 h. The reaction was quenched with water (50 mL), and the aq phase was extracted with CH2Cl2 (3 × 30 mL). The combined organic phases were dried over Na2SO4 and evaporated. The crude was purified by silica gel column chromatography (hexane/AcOEt 90/10→70/30) to obtain permethylated sugar 2 as yellow oil (2.172 g, 8.7 mmol, 56%). R f = 0.4 (hexane/AcOEt 70/30 v/v). The obtained data were consistent with those previously reported [20, 61-63]. IR (film) nmax 2910, 2829, 1450, 1359, 1200, 1098, 1052, 954 cm-1. 1H NMR (CDCl3, 500 MHz) δ 4.87 (1H, d, 3 J 1,2 = 3.7 Hz, H-1), 3.85 (1H, ddd, J 5,6 = 7.0, J 5,6 = 6.6, 3 J 5,4 = 1.0 Hz, H-5), 3.69 (1H, dd, J 4,3 = 3.0, J 4,5 = 1.0 Hz, H-4), 3.64 (1H, dd, J 2,3 = 10.0, J 2,1 = 3.7 Hz, H-2), 3.57 (3H, s, OMe-4), 3.56 (1H, dd, J 6,6 = 9.5, J 6,5 = 6.6 Hz, H-6), 3.54 (1H, dd, J 3,2 = 10.0, J 3,4 = 3.0 Hz, H-3), 3.520 (1H, dd, J 6,6 = 9.5, J 6,5 = 7.0 Hz, H-6), 3.518 (3H, s, OMe-2), 3.516 (3H, s, OMe-3), 3.42 (3H, s, OMe-1), 3.41 (3H, s, OMe-6). 13C NMR (CDCl3, 125 MHz) δ 98.1 (C1), 80.5 (C3), 78.0 (C2), 76.3 (C4), 71.4 (C6), 69.0 (C5), 61.5 (OMe-4), 59.3 (OMe-6), 59.1 (OMe-3), 58.3 (OMe-2), 55.4 (OMe-1). FAB+-MS m/z (rel. int.): 251 [M+H]+ (24), 219 (56), 187 (100), 154 (35), 145 (23). Anal. C 52.35, H 8.8, calcd for C11H22O6, C 52.8, H 8.9.
2,3,4,6-Tetra-O-methyl-a,b-D-galactose (3). In a round bottom flask with a Dean-Stark trap, compound 2 (1.5 g, 6.0 mmol) was dissolved in aq 2 N HCl (30 mL, 60.0 mmol). The mixture was heated to 70 °C for 3 h. The reaction mixture was neutralized with solid NaHCO3 and extracted with CH2Cl2 (6 × 30 mL). The combined organic extracts were dried over Na2SO4 and evaporated. The crude mixture of a and b epimers was purified by silica gel column chromatography (hexane/AcOEt 30/70 v/v) to yield hydrolyzed product 3 as colourless oil in a:b ≈ 3:1 ratio (0.767 g, 3.2 mmol, 54%). R f = 0.1 (hexane/AcOEt 30/70 v/v). [a]D = +65.87 (c 0.008, CH3CN). The obtained data were consistent with those previously reported [62]. IR (film) nmax 3399, 2930, 2833, 1451, 1367, 1199, 1064, 982, 951 cm-1. 1H NMR (CDCl3, 500 MHz) δ 5.40 (1H, d, J 1,2 = 3.6 Hz, H-1a), 4.55 (1H, d, J 1,2 = 7.5 Hz, H-1b), 4.15-4.11 (1H, m, H-5a), 3.70 (1H, dd, J 4,3 = 2.8, J 4,5 = 1.0 Hz, H-4a), 3.64 (1H, dd, J 2,3 = 7.0, J 2,1 = 3.6 Hz, H-2a), 3.63 (3H, s, OMe-b), 3.61 (1H, d, J 4,5 = 3.6 Hz, H-4b), 3.57-3.51 (6H, m, H-3a, H-5b, H-6a, H-6b), 3.565 (3H, s, OMe-a), 3.56 (3H, s, OMe-b) 3.527 (3H, s, OMe-a), 3.525 (3H, s, OMe-a), 3.52 (3H, s, OMe-b), 3.395 (3H, s, OMe-a), 3.392 (3H, s, OMe-b), 3.29 (1H, dd, J 2,3 = 9.7, J 2,1 = 7.5 Hz, H-2b), 3.17 (dd, J 3,2 = 9.6, J 3,4 = 3.1 Hz, H-3b). 13C NMR (CDCl3, 125 MHz) δ 97.45 (C1-b), 90.9 (C1-a), 83.9 (C3-b), 81.9(C2-b) 79.9 (C3(a), 78.0 (C2-a), 76.1 (C4-a), 75.0 (C4-b), 73.1 (C5-b), 71.4 (C6-a), 71.0 (C6-b), 69.0 (C5-a), 61.21 (OMe-a), 61.16 (OMe-b), 60.7 (OMe-b), 59.1 (OMe-a,b), 58.8 (OMe-a), 58.1 (OMe-b), 58.0 (OMe-a). FAB+-MS m/z (rel. int.): 237 [M+H]+ (10), 219 (68), 187 (100), 154 (22), 111 (39), 101 (88).
Acetyl 2,3,4,6-tetra-O-methyl-b-D-galactopyranoside (4b). In a round bottom flask sodium acetate (0.25 g, 3.0 mmol) was dissolved in acetic anhydride (5.5 mL, 19 mmol) and heated to 70 °C for 20 min. Compound 3 (0.730 g, 3.1 mmol) was added and the reaction mixture was heated to the same temperature for additional 20 min. The mixture was quenched with 20 mL aq soln 2 N NaHCO3 and extracted with CH2Cl2 (3 x 30 mL). The combined organic phases were dried over Na2SO4, filtered and evaporated. The crude mixture of a and b epimers was obtained (0.250 g, 0.9 mmol, 60%), a:b = 1:3 ratio. The mixture was separated by silica gel column chromatography (hexane/AcOEt 50/50 v/v) to yield 4b as a white solid and 4a as a transparent liquid. 4b R f = 0.6 (hexane/AcOEt 30/70 v/v). [a]D = +3.1 (c 0.008, CH3CN). IR (KBr) nmax 2918, 2823, 1753, 1225, 1101, 1075, 1040, 948 cm-1. 1H NMR (CDCl3, 500 MHz) δ 5.44 (1H, d, J 1,2 = 8.0 Hz, H-1), 3.70 (1H, dd, J 4,3 = 3.0, J 4,5 = 0.9 Hz, H-4), 3.63 (1H, ddd, J 5,6a = 7.8, J 5,6b = 5.3, J 5,4 = 0.9 Hz, H-5), 3.58 (1H, dd, J 6a,6b = 9.0, J 6a,5 = 7.8 Hz, H-6a), 3.58 (3H, s, OMe-4), 3.55 (3H, s, OMe-2), 3.54 (3H, s, OMe-3), 3.51 (1H, dd, J 6b,6a = 9.0, J 6b,5 = 5.3 Hz, H-6b), 3.48 (1H, dd, J 2,3 = 9.7, J 2,1 = 8.0 Hz, H-2), 3.38 (3H, s, OMe-6), 3.23 (1H, dd, J 3,2 = 9.7, J 3,4 = 3.0 Hz, H-3), 2.12 (3H, s, OAc-1). 13C NMR (CDCl3, 125 MHz) δ 169.5 (C=O), 94.4 (C1), 84.1 (C3), 79.5 (C2), 74.5 (C4), 73.8 (C5), 70.1 (C6), 61.4 (OMe-4), 61.0 (OMe-2), 59.3 (OMe-6), 58.4 (OMe-3), 21.2 (COMe-1). FAB+-MS m/z (rel. int.): 277 [M-H+] (5), 219 (52), 187 (100), 154 (23), 137 (24), 111 (59), 101 (98). Anal. C 51.8, H 8.0, calcd for C12H22O7, C 51.8, H 8.0.
Acetyl 2,3,4,6-tetra-O-methyl-α-D-galactopyranoside (4a) Transparent liquid. R f = 0.5 (hexane/AcOEt 50/50 v/v) 1H NMR (CDCl3, 400 MHz) δ 6.37 (1H, d, J 1,2 = 3.7 Hz, H-1), 3.96 (1H, ddd, J 5,6a = 7.5, J 5,6b = 5.7, J 5,4 = 0.7 Hz, H-5), 3.77 (1H, dd, J 4,3 = 2.8, J 4,5 = 1.0 Hz, H-4), 3.73 (1H, dd, J 2,3 = 10.2, J 2,1 = 3.7 Hz, H-2), 3.58 (3H, s, OMe-4), 3.55 (1H, dd, J 6a,6b = 9.3, J 6a,5 = 7.5 Hz, H-6a), 3.54 (3H, s, OMe-3), 3.52 (1H, dd, J 3,2 = 10.2, J 3,4 = 2.8 Hz, H-3), 3.48 (1H, dd, J 6b,6a = 9.3, J 6b,5 = 5.7 Hz, H-6b), 3.47 (3H, s, OMe-2), 3.39 (3H, s, OMe-6), 2.12 (3H, s, OMe-1). 13C NMR (CDCl3, 100 MHz) δ 169.6 (C=O), 90.1 (C1), 80.0 (C3), 77.0 (C2), 75.4 (C4), 71.6 (C5), 70.7 (C6), 61.5 (OMe-4), 59.3 (OMe-6), 59.2 (OMe-2), 58.2 (OMe-3), 21.2 (COMe-1). Anal. C 51.65, H 8.0, calcd for C12H22O7, C 51.8, H 8.0.
Synthesis of 2-methoxypyridine receptors 9-11
General Procedure A. In a three neck round-bottom flask 6-bromo-2-methoxypyridine 5 was dissolved in freshly distilled dry Et3N. The mixture was degassed bubbling N2 for 40 min. Then Pd(PPh3)2Cl2 catalyst and CuI were added. After 30 min, a solution of the corresponding alkyne in freshly distilled dry Et3N was added through cannula. The reaction mixture was left stirring at rt for 16 h. The reaction mixture was quenched with a saturated NH4Cl aqueous solution. The aqueous phase was extracted 3 times with AcOEt, washed with water and brine. The organic extracts were dried over Na2SO4 and evaporated.
6-(3-Phenylpropyn-1-yl)-2-methoxypyridine (9). Prepared according to general procedure A, 3-phenyl-1-propyne 6 (0.996 g, 6.0 mmol); Pd(PPh3)2Cl2 (0.225 g, 0.3 mmol, 0.05 eq.) and CuI (0.075 g, 0.367 mmol, 0.06 eq.) were dissolved in freshly distilled dry Et3N (30 mL). Starting material 6-bromo-2-methoxypyridine 5 (1.162 g, 6.0 mmol) was dissolved in degassed Et3N (10 mL). The crude was purified by silica gel column chromatography (hexane/AcOEt 98/02 v/v) to yield receptor 9 as yellow oil (1.179 g, 5.28 mmol, 88%). R f = 0.28 (hexane/AcOEt 95/05 v/v). IR (film) nmax 2947, 2228, 1568,1458, 1238, 1049, 799,729 cm-1. 1H NMR (CDCl3, 500 MHz) δ 7.48 (1H, dd, J 4,5 = 8.3, J 4,3 = 7.3 Hz, H-4), 7.44−7.39 (2H, m, Ar-Hortho), 7.36−7.31 (2H, m, Ar-Hmeta), 7.29−7.21 (2H, m, Ar-Hpara), 7.03 (1H, d, J 3,4 = 7.3 Hz, H-3), 6.67 (1H, d, J 5,4 = 8.3 Hz, H-5), 3.95 (3H, s, OMe-2), 3.86 (2H, s, CH2). 13C NMR (CDCl3, 125 MHz) δ 163.9 (C2), 140.6 (C6), 138.5 (C4), 136.2 (Cipso), 128.7 (Cortho), 128.2 (Cmeta), 126.9 (Cpara), 120.6 (C3), 110.9 (C5), 87.6 (C≡CCH2), 82.5 (Pyr-C≡C), 53.7 (OCH3), 25.9 (CH2). DART-MS m/z 225, 224 [M+H]+. HRMS (ESI+) m/z 224.1072 (calcd for C15H14NO [M+H]+: 224.1075).
6-(3-Cyclohexyl-propyn-1-yl)-2-methoxypyridine (10). Prepared according to general procedure A, 3-cyclohexyl-1-propyne 7 (0.8 mL, 6.0 mmol); Pd(PPh3)2Cl2 (0.225 g, 0.3 mmol, 0.05 eq.) and CuI (0.111 g, 0.6 mmol, 0.1 eq.) were dissolved in freshly distilled dry Et3N (30 mL). Starting material 6-bromo-2-methoxypyridine 5 (1.162 g, 6.0 mmol) was dissolved in degassed Et3N (10 mL). The crude was purified by silica gel column chromatography (hexane/AcOEt 97/03 v/v) to yield receptor 10 as yellow oil (0.770 g, 3.36 mmol, 56%). R f = 0.4 (hexane/AcOEt 95/05 v/v). IR (film) nmax 2921, 2848, 2226, 1568, 1459, 1238, 1048, 799, 729 cm-1. 1H NMR (CDCl3, 500 MHz) δ 7.47 (1H, dd, J 4,5 = 8.3, J 4,3 = 7.3 Hz, H-4), 6.99 (1H, dd, J 3,4 = 7.3, J 3,5 = 0.7 Hz, H-3), 6.65 (1H, dd, J 5,4 = 8.3, J 5,3 = 0.7 Hz, H-5), 3.94 (3H, s, OMe-2), 2.33 (2H, d, J CH2,CyC(1)H = 6.8 Hz, CH2), 1.98−1.82 (2H, m), 1.74 (2H, dt, J = 12.8, 3.3 Hz, Cy), 1.67 (1H, dddt, J = 12.8, 5.2, 3.3, 1.7 Hz, H-1’), 1.60 (1H, dddq, J = 13.9, 10.3, 6.8, 3.3 Hz, Cy), 1.27 (2H, qt, J = 12.8, 3.3 Hz, Cy), 1.16 (1H, qt, J = 12.7, 3.3 Hz, Cy), 1.06 (2H, qd, J = 12.8, 3.3 Hz, Cy). 13C NMR (CDCl3, 125 MHz) δ 163.9 (C2), 141.1 (C6), 138.5 (C4), 120.5 (C3), 110.5 (C5), 89.7 (C≡CCH2), 81.5 (Pyr-C≡C), 53.6 (OCH3), 37.5 (Cy), 33.0 (Cy), 27.4 (Cy), 26.4 (Cy), 26.3 (Cy). DART-MS m/z 231, 230 [M+H]+. HRMS (ESI+) m/z 230.1545 (calcd for C15H14NO [M+H]+: 224.1545).
6-(4-Phenylbutyn-1-yl)-2-methoxypyridine (11). Prepared according to general procedure A, 4-phenyl-1-butyne 8 (1.82 g, 7.0 mmol); Pd(PPh3)2Cl2 (0.640 g, 0.7 mmol, 0.1 eq.) and CuI (0.152 g, 0.8 mmol, 0.11 eq.) were dissolved in freshly distilled dry Et3N (40 mL). Starting material 6-bromo-2-methoxypyridine 5 (1.357 g, 7.0 mmol) was dissolved in degassed Et3N (10 mL). The crude was purified by silica gel column chromatography (hexane/AcOEt 99/01 v/v) to yield receptor 11 as a yellow oil (0.864 g, 3.64 mmol, 52%). R f = 0.33 (hexane/AcOEt 99/01 v/v). IR (film) nmax 2946, 2228, 1569, 1459, 1240, 1052, 800, 731 cm-1. 1H NMR (CDCl3, 500 MHz): 7.47 (1H, dd, J 4,5 = 8.4, J 4,3 = 7.3 Hz, H-4), 7.33−7.20 (5H, m, Ar), 6.95 (1H, dd, J 3,4 = 7.3, J 3,5 = 0.7 Hz, H-3), 6.66 (1H, dd, J 5,4 = 8.4, J 5,3 = 0.7 Hz, H-5), 3.94 (3H, s, OMe-2), 2.95 (2H, t, J CH2,CH2 = 7.7 Hz, -CH2Ph), 2.72 (2H, t, J CH2,CH2 = 7.7 Hz, ≡CCH2). 13C NMR (CDCl3, 125 MHz) δ 163.9 (C2), 140.8 (Cipso), 140.6 (C6), 138.5 (C4), 128.63 (Cmeta), 128.56 (Corto), 126.5 (Cpara), 120.4 (C3), 110.7 (C5), 89.6 (C≡CCH2), 81.2 (Pyr-C≡C), 53.6 (OCH3), 35.0 (CH2), 21.9 (CH2). DART-MS m/z 239, 238 [M+H]+. HRMS (ESI+) m/z 238.1230 (calcd for C16H16NO [M+H]+: 238.1232).
Synthesis of 2(1H)-pyridone receptors 12-14
General Procedure B. In a two neck round bottom flask the obtained product in general procedure A (1.0 eq.) was dissolved in freshly distilled dry acetonitrile. The reaction mixture was degassed bubbling N2 for 40 minutes, then NaI (1.0 eq.) was added and TMSCl (1.0 eq.) was added dropwise with a glass syringe. The reaction was left stirring for 3 days at rt. The mixture was quenched adding Na2S2O3 aqueous solution (1.0 M). The aqueous phase was extracted three times with AcOEt, washed with water and brine. The organic extracts were dried over Na2SO4 and evaporated.
6-(3-Phenyl-propyn-1-yl)-2(1H)-pyridone (12). Prepared according to general procedure B, compound 9 (0.893 g, 4.0 mmol) was dissolved in freshly distilled dry acetonitrile (10 mL); NaI (0.435 g, 4.0 mmol); TMSCl (0.5 mL, 4.0 mmol). The crude was purified by solvent pair recrystallization to yield yellow crystals of target product 12 (0.276 g, 1.32 mmol, 33%). mp 85−88 °C (CH2Cl2/hexane). R f = 0.2 (hexane/AcOEt 50/50 v/v). IR (KBr) nmax 2784, 2236, 1642, 1544, 1447, 795, 732 cm-1. 1H NMR (CDCl3, 500 MHz) δ 12.40 (1H, s, NH), 7.43−7.40 (2H, m, Ar), 7.36−7.32 (1H, m, Ar), 7.353 (1H, dd, J 4,5 = 9.2, J 4,3 = 6.9 Hz, H-4) 7.28−7.24 (1H, m, Ar), 6.58 (1H, dd, J 3,4 = 9.2, J 3,5 = 1.0 Hz, H-3), 6.38 (1H, dd, J 5,4 = 6.9, J 5,3 = 1.0 Hz, H-5), 3.86 (2H, s, CH2). 13C NMR (CDCl3, 125 MHz) δ 164.8 (C=O), 140.9 (C4), 135.3 (Cipso), 129.8 (C6), 128.8 (Ar), 128.2 (Ar), 127.1 (Ar), 120.8 (C3), 111.3 (C5), 93.9 (≡C-CH2), 76.0 (Pyr-C≡), 25.9 (CH2). DART-MS m/z 210 [M+H]+. HRMS (ESI+) m/z 210.0917 (calcd for C14H12NO [M+H]+: 210.0919).
6-(3-Cyclohexyl-propyn-1-yl)-2(1H)-pyridone (13). Prepared according to general procedure B, compound 10 (0.459 g, 2.0 mmol,) was dissolved in freshly distilled dry acetonitrile (10 mL); NaI (0.218 g, 2.0 mmol); TMSCl (0.250 mL, 2.0 mmol). The crude was purified by silica gel column chromatography (hexane/AcOEt 60/40 v/v) to yield compound 13 as yellow crystalline solid (0.202 g, 0.94 mmol, 47%). mp 110 °C. R f = 0.4 (hexane/AcOEt 60/40 v/v). IR (KBr) nmax 3291, 2920, 2850, 2227, 1765, 1647, 1544, 1447, 794, 721 cm-1. 1H NMR (CDCl3, 500 MHz) δ 11.76 (1H, s, NH), 7.33 (1H, dd, J 4,3 = 9.2, J 4,5 = 6.9 Hz, H-4), 6.53 (1H, dd, J 3,4 = 9.2, J 3,5 = 1.0 Hz, H-3), 6.32 (1H, dd, J 5,4 = 6.9, J 5,3 = 1.0 Hz, H-5), 2.32 (2H, d, J CH2,CyC(1)H = 6.7 Hz, CH2), 1.91−1.81 (2H, m, Cy), 1.74 (2H, dt, J = 12.7, 3.3 Hz, Cy), 1.67 (1H, dddq, J = 11.8, 5.2, 3.6, 1.7 Hz, H-1’), 1.59 (1H, tdp, J = 13.7, 6.7, 3.6 Hz, Cy), 1.27 (2H, qt, J = 12.7, 3.3 Hz, Cy), 1.17 (1H, qt, J = 12.7, 3.3 Hz, Cy), 1.06 (2H, qd, J = 12.7, 3.3 Hz, Cy). 13C NMR (CDCl3, 125 MHz) δ 164.6 (C=O), 140.9 (C4), 130.2 (C6), 120.4 (C3), 110.9 (C5), 95.9 (≡C-CH2), 75.1 (Pyr-C≡), 37.2 (Cy), 32.8 (Cy), 27.3 (Cy), 26.3 (Cy), 26.2 (Cy). DART-MS m/z 216 [M+H]+. HRMS (ESI+) m/z 216.1388 (calcd for C14H18NO [M+H]+: 216.1388). Anal. C 77.9, H 7.5, N 6.5, calcd for C14H17NO, C 78.1, H 8.0, N 6.5.
6-(4-Phenylbutyn-1-yl)-2(1H)-pyridone (14). Prepared according to general procedure B, compound 11 (0.830 g, 3.5 mmol) was dissolved in freshly distilled dry acetonitrile (20 mL); NaI (0.525 g, 3.5 mmol); TMSCl (0.444 mL, 10.5 mmol). The crude was purified by silica gel column chromatography (hexane/AcOEt 50/50 v/v) to yield compound 14 as colorless crystals (0.367 g, 1.64 mmol, 47%). mp 139 °C. R f = 0.16 (hexane/AcOEt 30/70 v/v). IR (KBr) nmax 2780, 2227, 1633, 1543, 1452, 796, 718 cm-1. 1H NMR (CDCl3, 500 MHz) δ 11.76 (1H, s, NH), 7.32 (1H, dd, J 4,3 = 9.2, J 4,5 = 6.9 Hz, H-4), 7.32-7.29 (2H, m, Hortho), 7.27-7.25 (2H, m, Hmeta), 7.24-7.21 (1H, m, Hpara), 6.56 (1H, dd, J 3,4 = 9.2, J 3,5 = 1.0 Hz, H-3), 6.29 (1H, dd, J 5,4 = 6.9, J 5,3 = 1.0 Hz, H-5), 2.93 (2H, t, J CH2,CH2 = 7.5 Hz, -CH2Ph), 2.72 (2H, t, J CH2,CH2 = 7.5 Hz, ≡CCH2-).13C NMR (CDCl3, 125 MHz) δ 164.7 (C=O), 140.9 (C4), 140.2 (Cipso), 129.9 (C6), 128.6 (Cortho, Cmeta), 126.6 (Cpara), 120.7 (C3), 111.1 (C5), 95.8 (≡C-CH2), 74.9 (Pyr-C≡), 34.5 (-CH2Ph), 21.8 (≡C-CH2). EI-MS m/z (rel. int.): 224 (14), 223 [M]+ (67), 222 (36), 91 (100). HRMS (ESI+) m/z 224.1075 (calcd for C15H14NO [M+H]+: 224.1075). Anal. C 79.9, H 5.8, N 6.2, calcd for C15H13NO, C 80.7, H 5.9, N 6.3.

