











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El síndrome de Stevens-Johnson, en conjunto con la necrólisis epidérmica tóxica son alteraciones que forman parte de las reacciones medicamentosas severas. Ambas se caracterizan por lesiones cutáneas y mucosas similares, que varían según la evolución de la enfermedad e incluyen maculas eritematosas, en ocasiones de configuración en diana, y posteriormente eritema generalizado y formación de ampollas flácidas en la piel, mientras que en las mucosas la manifestación principal son las erosiones. Se diferencian según la extensión de la afección, considerándose síndrome de Stevens-Johnson a la afección menor del 10% de la superficie corporal, y la mortalidad estimada es del 5%.6 Estas afecciones pueden provocar morbilidad y mortalidad importante, usualmente paralela al daño de la superficie corporal.
INFORME DE CASO
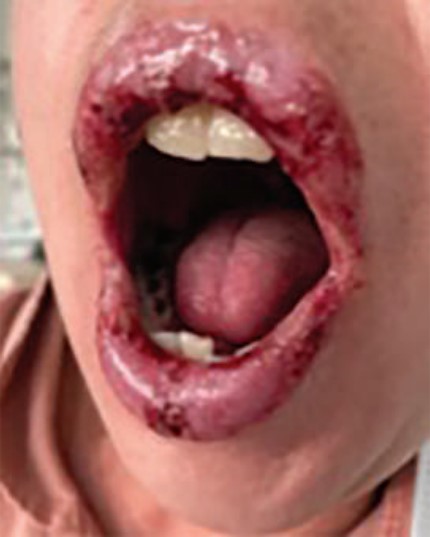
Paciente femenina de 40 años, con antecedentes médicos de endometriosis y colitis ulcerativa crónica inflamatoria de reciente diagnóstico, en tratamiento con sulfasalazina, a dosis de 1000 mg/tres veces al día, daflazacort de 30 mg/día y anticonceptivos orales. Fue enviada al servicio de Alergología para internamiento por sospecha de reacción medicamentosa asociada con sulfasalazina, que había iniciado 20 días previos al inicio de la dermatosis, caracterizada por exantema maculopapular eritematoso pruriginoso en la cara, el tronco y las extremidades superiores, con posterior afectación y erosiones dolorosas en la mucosa oral y vaginal, queilitis y eritema ocular. Se inició tratamiento con metilprednisolona, a dosis de 125 mg/día por vía intravenosa durante tres días, seguido de corticoesteroides orales, además de fluidoterapia y tratamiento tópico con corticoesteroides y emolientes. No se documentó fiebre, eosinofilia, lesión renal, trastorno hidroelectrolítico ni datos de sobreinfección. Con el tratamiento descrito hubo mejoría clínica. Puesto que la afección fue menor del 10% de la superficie corporal total, se estableció el diagnóstico de síndrome de Stevens-Johnson. Se decidió de forma interdisciplinaria que la paciente no podía recibir sulfamidas ni medicamentos asociados con reacción cruzada con éstos fármacos, por lo que se inició tratamiento con azatioprina, a dosis de 2 mg/kg para el trastorno intestinal.
Posterior al egreso hospitalario se citó para efectuar la prueba de transformación linfocitaria con sulfasalazina, que resultó positiva. Figura 1 y Figura 2

DISCUSIÓN
El síndrome de Stevens-Johnson es una reacción medicamentosa poco frecuente, que puede aparecer en personas de cualquier edad, género o raza, que consumen algún fármaco desencadenante del evento de hipersensibilidad. La incidencia anual estimada es de 1.2-6 por cada 1,000,000 de personas y la mortalidad del 5%. La principal causa de muerte es la sepsis.1
Al analizar la distribución por edad, similar a lo encontrado en estudios de anafilaxia en América latina, en este ensayo encontramos que el 56.5% de los pacientes tenía entre 19-60 años. En el estudio OLASA se identificó que, de los 534 pacientes incluidos, la mayor proporción de casos eran mayores de18 años, 42% tenía entre 18-40 años y el 26.5% pertenecía al grupo de mayores de 40 años.8
El mecanismo exacto por el que se produce la reacción aún se desconoce, pero puede asociarse con una reacción de hipersensibilidad tipo IV. Incluso se ha relacionado con alguna alteración para la degradación productos tóxicos de los medicamentos, que resulta en apoptosis de los queratinocitos, quizá mediada por TNF-α, proteína FAS y granzima B.1,2
Además, existe una respuesta inmunitaria por parte de los linfocitos CD8+ citotóxicos, con liberación de citoquinas que generan una respuesta inflamatoria.
Los fármacos son los agentes causales más frecuentes del síndrome de Stevens-Johnson; sin embargo, pueden estar implicados agentes infecciosos, principalmente Mycoplasma pneumoniae. En el 15-30% de los casos no puede identificarse el agente causal.3 Cuadro 1
De acuerdo con las manifestaciones de la enfermedad, se clasifica según el porcentaje de área de superficie corporal afectada. Se denomina síndrome de Stevens-Johnson cuando existe afectación menor del 10% de la superficie corporal.4
En cuanto a las manifestaciones clínicas, puede haber una fase prodrómica con fiebre y malestar general. Posteriormente aparecen lesiones cutáneas, usualmente máculas eritematosas o lesiones en diana atípicas en el tronco, que tienden a confluir, con posterior aparición de ampollas flácidas y necrosis epidérmica. El 80% de los pacientes pueden tener lesiones en las mucosas, sobre todo en la oral, con ulceración y erosiones. También puede haber afectación de la conjuntiva ocular y la mucosa genital.5
La escala de SCORTEN se utiliza como predictor de mortalidad. El resultado varía de 0 a 7 puntos, y el puntaje más alto se asocia con mayor mortalidad.6
En este estudio, el agente causal más frecuente de anafilaxia fueron los fármacos (52.1%), sobre todo los AINEs, en pacientes mayores de 60 años. En el estudio OLASA se observó un comportamiento similar con los AINE's, que se encuentran implicados frecuentemente en eventos de anafilaxia; sin embargo, los pacientes con mayor afectación fueron los mayores de 40 años.8
En la paciente del caso aquí expuesto, la escala de SCORTEN fue de 1 punto, correspondiente con la edad, para una mortalidad asociada baja. Cuadro 2
Para establecer el diagnóstico de síndrome de Stevens-Johnson debe considerarse la morfología y extensión de las lesiones, además del medicamento consumido o infecciones preexistente, que llevan a la sospecha clínica. Se ha descrito un periodo de 4 a 28 días entre el inicio del fármaco y la aparición de la reacción medicamentosa, pero puede ocurrir, incluso, dos meses después.7 Este dato coincide con nuestro caso clínico, que tuvo un intervalo de 20 días desde el inicio del fármaco hasta la manifestación de las lesiones.
La histopatología se utiliza para confirmar el diagnóstico y descartar otras enfermedades. El reporte suele indicar queratinocitos necróticos, cambios vacuolares basales, ampollas subepidérmicas, infiltrado inflamatorio denso en la dermis y extravasación de eritrocitos.7 En cuanto a la biopsia de la paciente, se obtuvo de forma tardía, del labio inferior, cuando había recibido tratamiento. El estudio informó: zona ulcerada en la epidermis, queratinocitos con cambios reactivos e infiltrado inflamatorio compuesto por linfocitos y macrófagos. Figura 3
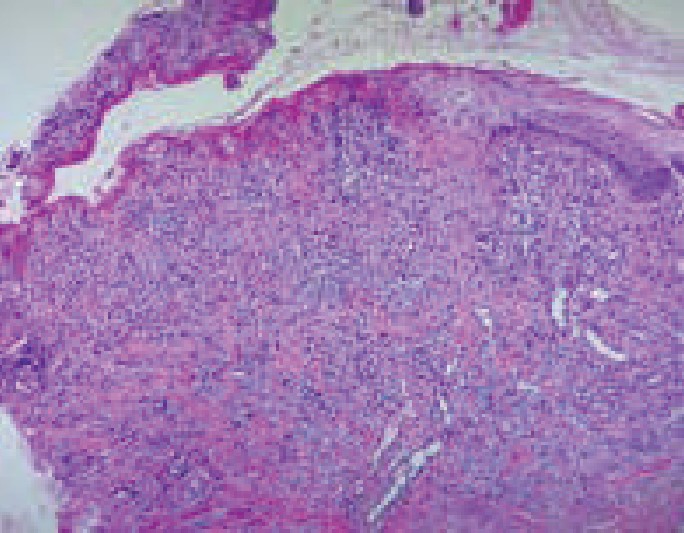
Figura 3. Zona ulcerada que muestra, en el fondo de la lesión, infiltrado inflamatorio linfoide y proliferación vascular (100 X).
Para confirmar el diagnóstico se llevó a cabo la prueba de transformación linfocitaria, utilizada para reacciones no inmediatas o mediadas por linfocitos T. La prueba se basa en la estimulación de los linfocitos por el alérgeno y la incorporación de timidina tritiada al ADN, con posterior medición de la 3H-timidina en las muestras de cultivo mediante la detección de la radiación.8,9
El resultado fue positivo y s confirmó la sospecha inicial de sulfamida como agente causal de la reacción medicamentosa severa. Cuadro 3
Respecto al tratamiento del síndrome de Stevens-Johnson, deben considerarse las siguientes medidas:10-12
Medidas de soporte. Identificación y retiro del medicamento causal. Los pacientes deben recibir tratamiento en una unidad de cuidados intensivos, con evaluación de la vía aérea, función renal, electrolitos, nutrición, control del dolor y prevención de infecciones.
Cuidados de las lesiones. Algunos centros realizan desbridamiento quirúrgico y otros prefieren dejar la piel desprendida como apósito biológico; ambas opciones han demostrado tasas similares de reepitelización. Puede utilizarse una gasa embebida con vaselina o algún ungüento que contenga plata, incluso se han utilizado sustitutos sintéticos de piel.
Fluidoterapia y nutrición. Las opciones recomendadas son electrolitos o soluciones con albumina
Terapia coadyuvante:
Corticoesteroides sistémicos: su prescripción sigue discutiéndose; los estudios recientes no han logrado demostrar ventajas acerca de su indicación versus medidas de soporte.
Inmunoglobulina intravenosa: se administra frecuentemente en casos severos; sin embargo, no demostrado beneficio significativo en la supervivencia.
Inhibidores de TNF-α: existen reportes de casos tratados con etanercept e infliximab. Es posible que puedan detener la progresión de la necrosis epidérmica y contribuir con la reepitelización.
Ciclosporina: Algunos estudios indican que disminuye la evolución del síndrome de Stevens-Johnson y aumenta la tasa de supervivencia.
Plasmaferesis: los casos reportados con este tratamiento no han informado mejoría significativa en cuanto a reepitelización ni mortalidad.
Cuadro 1 Fármacos comúnmente asociados con el síndrome de Steven-Johnson/necrólisis epidérmica tóxica4
| Anticonvulsivantes | Lamotrigina |
| Fenitoina | |
| Carbamazepina | |
| Antibióticos | Trimetoprima sulfametoxasol |
| Penicilinas | |
| Tetraciclinas | |
| Cefalosporinas | |
| AINES | |
| Alopurinol | |
| Nevirapina | |
| Sulfamidas | |
| Antiparasitarios | |
Cuadro 2 Escala SCORTEN
| Variablesa | Valores | Puntaje |
|---|---|---|
| Edad | ≥ 40 años | 1 |
| Frecuencia cardiaca | ≥ 120 lpm | 1 |
| Datos de malignidad | Cáncer o neoplasia hematológica | 1 |
| Epidermólisis inicial | ≥ 10% de la superficie corporal afectada | 1 |
| Nitrógeno ureico sérico | ≥ 10 mmol/L (27 mg/dL) | 1 |
| Bicarbonato sérico | < 20 mmol/L (20 mEq/L) | 1 |
| Glucosa sérica | ≥ 14 mmol/L (252 mg/dL) | 1 |
CONCLUSIONES
El síndrome de Stevens-Johnson es una reacción asociada con lesiones cutáneas, potencialmente mortal, frecuentemente provocada por el consumo de ciertos fármacos. La prueba de transformación linfocitaria es un método útil que puede confirmar el agente causal y prevenir complicaciones importantes a futuro.

