










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versão impressa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.65 no.3 México Mai./Jun. 2008
Caso clínico
Secuelas neurológicas en tres pacientes con fenilcetonuria clásica diagnosticada tardíamente
Neurological sequelae in three patients with late-diagnosed classic phenylketonuria
Alejandra Sánchez-Peña1, Laura Martínez-de Villarreal1, Georgina Arteaga-Alcaraz1, Rosario Torres-Sepúlveda1, Alma Rosa Marroquín-Escamilla2, Valdemar Abrego-Moya2, Zacarías Villarreal-Pérez3, Carmen Esmer-Sánchez1
1 Departamento de Genética, Facultad de Medicina, Hospital Universitario Dr. José E. González, Universidad Autónoma de Nuevo León, Monterrey, México.
2 Departamento de Pediatría, Hospital Universitario Dr. José E. González, Universidad Autónoma de Nuevo León, Monterrey, México.
3 Departamento de Endocrinología, Hospital Universitario Dr. José E. González, Universidad Autónoma de Nuevo León, Monterrey, México.
Solicitud de sobretiros:
Dra. Laura Martínez de Villarreal,
Departamento de Genética, Facultad de Medicina,
Universidad Autónoma de Nuevo León,
Ave. Francisco I. Madero Esq. Eduardo Aguirre Pequeño
s/n, Col. Mitras Centro, C.P. 64460,
Monterrey, Nuevo León, México.
Fecha de recepción: 04-09-2007.
Fecha de aprobación: 10-04-2008.
Resumen
Introducción. La fenilcetonuria es un padecimiento que puede diagnosticarse oportunamente, sin embargo, aún es posible detectar pacientes con secuelas neurológicas graves que desafortunadamente no fueron sujetos a un estudio al nacimiento.
Casos clínicos. El primer caso se trató de una paciente de 12 meses que acudió por retardo en el desarrollo psicomotor, sus niveles de fenilalanina en sangre fueron de 1 285 μmol/L (normal 31-75 μmol/L). Su hermana mayor (caso 2) mostraba retardo en el desarrollo psicomotor considerado secundario a secuelas de encefalopatía hipóxico-isquémica, sin embargo también mostró hiperfenilalaninemia. El caso 3 se trató de una niña de 10 años que acudió por retardo psicomotor e hiperactividad; presentó ansiedad, irritabilidad, microcefalia, cabello rubio y piel clara. Los niveles de fenilalanina fueron de 1 170 μmol/L. En todos los casos se inició dieta baja en fenilalanina y después de un mes los niveles disminuyeron a la mitad. Actualmente mantienen valores normales de fenilalanina y muestran mejoría neurológica notable.
Conclusión. La intervención nutricional puede revertir algunas secuelas neurológicas en los casos con diagnóstico tardío de fenilcetonuria.
Palabras clave: Fenilalanina; fenilcetonuria; retardo mental; tamiz neonatal; manejo nutricional.
Abstract
Introduction. Phenylketonuria is a genetic disease that can be diagnosed easily and treated promptly avoiding long-term disabilities. Nevertheless, some children still lack neonatal screening as well as appropriate diagnosis, and they may present serious irreversible neurological damage.
Case report. We report 3 cases. Case 1 is a 12 month-old female with motor and developmental delay. Phenylalanine levéis were 1 285 μmol/L (normal values 31-75 μmol/L). Her sister (case 2) was a 6 year-old mentally retarded child previously thought to be due to hypoxic-ischemic encephalopathy; her phenylalanine levéis were 1 729 μmol/L. Case 3 describes a 10 year-old female with developmental delay, hyperactivity, anxiety, irritability, microcephaly, light-colored hair, and white skin. Phenylalanine levéis were 1170 μmol/L. A low-phenylalanine diet was prescribed for each patient. One month later they were evaluated and showed significantly reduced phenylalanine levéis (50%). Currently, they maintain normal values and show im-portant physical and neurological improvement.
Conclusions. In cases of late-diagnosed phenylketonuria, a prompt treatment with a strict nutritional management may revert some of the neurological damage developed in these patients.
Key words: Phenylketonuria; phenylalanine; neonatal screening; mental retardation; dietary restrictions.
Introducción
Los errores innatos del metabolismo son rasgos heredados que resultan de la ausencia o actividad reducida de una enzima o cofactor enzimático específico.1,3 La mayoría de estos defectos se acompañan de manifestaciones clínicas graves que a menudo aparecen poco después del nacimiento, siendo rápidamente evidente el retardo mental y la afección neurológica grave.4,7 Aunque individualmente los errores innatos del metabolismo son poco frecuentes, en conjunto no lo son; aproximadamente uno de cada 1 000 recién nacidos presentará uno de ellos.
El tamiz neonatal es un procedimiento para descubrir oportunamente aquellos recién nacidos, aparentemente sanos, que tienen una enfermedad que con el tiempo ocasionará daños graves e irreversibles. La detección oportuna de estos padecimientos permite tratarlos y evitar o aminorar sus consecuencias.8,11 El estudio se realiza en gotas de sangre capilar, usualmente obtenidas del talón y colectadas en papel filtro específico (también llamada ''tarjeta de Guthrie'').3 El tamiz neonatal ha sido muy efectivo para prevenir el retardo mental en pacientes con fenilcetonuria e hipotiroidismo congénito, entre otras enfermedades.2 En cada país, la legislación frente a la realización del estudio en recién nacidos es diferente. Esto tiene como consecuencia la existencia de casos en los que el diagnóstico se establece tardíamente cuando los pacientes muestran un grado de afección neurológica grave.13 Se presentan tres casos con diagnóstico tardío de fenilcetonuria clásica y sus principales consecuencias clínicas y neurológicas.
Presentación de los casos clínicos
Caso 1. Femenino de 12 meses de edad que acude a consulta pediátrica por sospecha de retardo psicomotor. La paciente es producto de la cuarta gesta, con antecedente de retardo psicomotor en una hermana mayor; embarazo y el parto sin complicaciones. A la exploración física se le encontró olor a moho, peso 8 500 g (p3), longitud 79 cm (p50), perímetro cefálico (PC) 41.5 cm (<p3), desnutrición de segundo grado, microcefalia, cabello hipopigmentado, pupilas mióticas con respuesta a la luz; piel seca y áspera. El examen neurológico muestra hipotonía, reflejos osteotendinosos disminuidos, mioclonías, movimientos estereotipados; prueba de Denver con desarrollo personal-social en tres meses, motor-adaptativo en cuatro meses, lenguaje en seis meses, motor-grueso seis meses.
Electroencefalograma (EEG) con brotes bilaterales de espiga-poliespiga y ondas lentas compatibles con actividad interictal de crisis generalizadas; potenciales evocados, resonancia magnética y perfil tiroideo normales. En el tamiz metabólico en orina las pruebas de cloruro férrico y Millón fueron positivas, y se observó fenilalanina y metionina en la cromatografía de capa fina. La cuantificación de aminoácidos en sangre por cromatografía de líquidos de alta resolución (HPLC) reportó niveles muy elevados de fenilalanina (1 285 μmol/L), estableciéndose el diagnóstico de fenilcetonuria clásica (PKU). Se inició intervención nutricional12 con una dieta baja en fenilalanina (300 mg/día), fórmula láctea especial (Lofenalac®) 200 mg/día con un contenido de proteínas de 30 g al día y complemento de tirosina (1 g/ día), la cantidad de energía fue prescrita de acuerdo a la edad, los carbohidratos y grasas en porcentajes de 50 y 35% del valor calórico total respectivamente.
A los dos meses postratamiento las crisis convulsivas disminuyeron, aumentó el contacto con el medio ambiente, mejoró la postura, el tono muscular, el cabello empezó a oscurecer, y los niveles en sangre de fenilalanina disminuyeron. Al tercer mes continuó la mejoría y los valores sanguíneos de fenilalanina se normalizaron. A seis meses de iniciado el tratamiento se recuperó el estado nutricional, somatometría en percentilas normales, su cabello se encuentra de color oscuro y sus valores sanguíneos de fenilalanina persisten normales. La valoración de Denver mostró desarrollo personal-social en 10 meses, motor-adaptativo en 11 meses, lenguaje en 10 meses, motor' grueso 12 meses (Fig. 1).
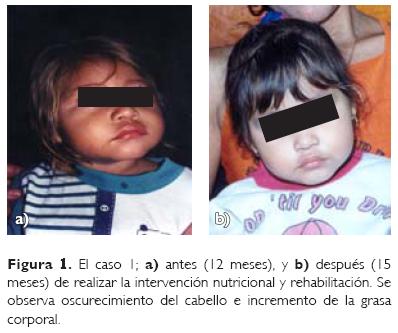
Caso 2. Se trató de la hermana mayor del caso 1. Esta paciente fue producto del tercer embarazo, resuelto por parto distócico; a los seis años de edad mostró retardo psicomotor catalogado como secundario a asfixia perinatal. A la exploración física presentaba pelo rubio, estrabismo, olor a moho, piel seca y áspera; neurológicamente con hiperactividad, movimientos repetitivos con rasgos autistas y su valoración de Denver mostró desarrollo personal-social en tres años dos meses, motor-adaptativo en dos años un mes, lenguaje en dos años, motor-grueso cuatro años cuatro meses. Se le realizó HPLC, el cual reportó niveles de fenilalanina en 1 729 μmol/L. Se inició intervención nutricional bajo los mismos parámetros que el caso 1, dieta baja en fenilalanina con 40 g de proteína al día, fórmula láctea especial (lofenelac®), complemento de tirosina.12
Al tercer mes mejoró su patrón conductual, su cabello se apreció más oscuro y los valores sanguíneos de fenilalanina disminuyeron. A los seis meses de tratamiento se refirió libre de crisis convulsivas y continuó con el oscurecimiento de cabello. Su valoración de Denver mostró desarrollo personal-social en tres años 10 meses, motor-adaptativo en cuatro años, lenguaje en dos años seis meses, motor-grueso cuatro años siete meses.
Caso 3. Femenina de 10 años de edad, primer producto de madre y padre de 19 años, sanos, no consanguíneos, con el antecedente de una hermana de cinco años sana. Embarazo con control prenatal regular, resuelto a término en medio hospitalario por parto distócico, con trabajo de parto prolongado y uso de fórceps, pesó 3 550 g y midió 51 cm. Acude por retardo psicomotor, hiperactividad y agresividad; espasmos del sollozo que iniciaron antes del año y remitieron a los cinco años de edad. La madre refiere mal olor de secreciones corporales. A la exploración física con peso de 27.2 kg (p10), talla 132 cm (p10), PC 49.7 cm (<p3), microcefalia, cabello rubio, piel clara, facies sin dismorfias, gingivitis, sin alteración cardiopulmonar, extremidades hipotróficas, tono y fuerza muscular adecuados. Neurológicamente con irritabilidad e hiperactividad, su valoración de Denver mostró desarrollo personal-social en tres años 10 meses, motor-adaptativo en cuatro años, lenguaje en dos años seis meses, motor-grueso cuatro años siete meses. Se detectó fenilalanina en 1 170 umol/L (30.4-120.5). Se inició dieta de 2 400 calorías diarias, con 40.5 g de proteínas (equivale a 140 mg/kg de fenilalanina), 3 g/día de tirosina, carbohidratos 50% y grasas 35% del valor calórico total; se suplemento ácido fólico 2.5 mg/día y selenio 100 mg/día.12 A los tres meses de tratamiento la paciente mejoró su estado general, aumentó de peso, su pelo se ha oscurecido, es menos agresiva, se encuentra más reactiva y su valoración de Denver mostró desarrollo personal-social en cuatro años dos meses, motor-adaptativo en cuatro años, lenguaje en tres años y motor-grueso cuatro años nueve meses.
Discusión
Actualmente se conoce que la detección oportuna y el tratamiento nutricional inmediato en los errores innatos del metabolismo conducen al desarrollo normal del neonato.9,11 En México es obligatorio realizar tamiz para hipotiroidismo congénito a todos los recién nacidos,2,14 mientras que el tamizaje para fenilcetonuria sólo se realiza en algunas instituciones privadas o de gobierno que cuentan con la infraestructura para hacerlo, de tal manera que no existe una cobertura nacional. Esto tiene como consecuencia, que todavía se presenten casos detectados tardíamente con problemas neurológicos graves. La fenilcetonuria (PKU, OMIM 261600) afecta a uno de cada 16 000 recién nacidos, se hereda en forma autosómica recesiva y ocurre por deficiencia de la enzima fenilalanina hidroxilasa (PAH). Cuando la enzima está deficiente la conversión de fenilalanina en tirosina se altera, con la consecuente disminución de las concentraciones de tirosina y aumento en las concentraciones de fenilalanina en plasma (por arriba de 120 μmol/L en ayunas) y en orina.1 El aumento en la concentración de fenilalanina causa un olor característico en la piel y orina, mientras que la deficiencia de tirosina afecta la síntesis de melanina, causando hipopigmentación de la piel y el pelo. También se afecta la síntesis de neurotransmisores como la dopamina, norepinefrina y serotonina y la formación de mielina, lo que resulta en retardo mental irreversible.7,15
El retardo mental aparece lentamente y puede pasar inadvertido durante unos meses, el único síntoma precoz de la enfermedad son los vómitos, a los que se les llegan a atribuir otras etiologías como reflujo gastroesofágico o estenosis pilórica. En otros casos, como los aquí descritos, no se sospechan hasta que se presentan las consecuencias neurológicas. Se calcula que un paciente que no ha sido tratado ha perdido alrededor de 50 puntos del cociente intelectual al cumplir el primer año. En los pacientes que no se detectan en el período neonatal se observan diversas manifestaciones neurológicas ocasionadas por la exposición crónica a niveles altos de fenilalanina; la deficiencia mental es la característica más importante de la fenilcetonuria no tratada, estos casos parecen normales al nacer y el retardo de su desarrollo intelectual puede pasar inadvertido un tiempo.16 Una tercera parte de los enfermos no presentan signos neurológicos, mientras que en otra tercera parte, los enfermos pueden sufrir parálisis cerebral grave. Además de la presencia de retardo mental con grados variables de afección, se ha informado la presencia de alteraciones psicológicas y conductuales, entre las cuales se encuentra hiperactividad, irritabilidad, trastornos del sueño, agitación psicomotriz, berrinches, ataques de rabia incontrolables, poca capacidad de atención, conducta agresiva, poca capacidad de aprendizaje, conductas psicóticas o autistas, y automutilación.17 Un 80% de los examinados presentan EEG anormal, con alteraciones múltiples, y alrededor de una cuarta parte de los enfermos sufren convulsiones. En la exploración neurológica, los hallazgos son inconstantes, pero la mayor parte tienen hipertonía y reflejos tendinosos exaltados (EEG). Muchos de estos hallazgos neurológicos se pueden catalogar como parálisis cerebral infantil, como ocurrió en la hermana del caso 1, quien erróneamente se diagnosticó como secuelas de hipoxia neonatal, esto llevó al nacimiento de otra hermana afectada. Ambas presentaban hipopigmentación cutánea y olor peculiar; las manifestaciones cutáneas como eccema y la hipopigmentación pueden manifestarse en forma precoz y deben hacer sospechar el diagnóstico. En familias con piel oscura, los enfermos son más rubios que sus hermanos no afectados, también el color de sus ojos es más claro y pueden llegar a ser azules. El olor de un enfermo fenilcetonúrico ha sido descrito como el de un ratón o rancio, y se ha correlacionado con la excreción por la orina de ácido fenilacético, es frecuente que los padres refieran este olor pero también es frecuente que el médico no enfoque su atención en ello; nuestros tres casos presentaron este síntoma por años antes de sospecharse el diagnóstico de fenilcetonuria.
En estos casos, el tamiz metabólico se realizó como parte del algoritmo diagnóstico de retardo mental, lo que refleja la necesidad de que en todo paciente con estas características se realice este estudio.
En la fenilcetonuria, la causa del retardo mental y de las manifestaciones neurológicas está siendo estudiada; se plantea que puede estar relacionada con un efecto tóxico del aumento de fenilalanina y sus metabolitos secundarios (fenilpirúvico, fenilláctico) en los líquidos corporales durante las primeras etapas de la vida.18 Por otro lado, algunos autores refieren que la escasa biodisponibilidad de tirosina para la síntesis de proteínas y neurotransmisores en el cerebro, es la causa de la afección neurológica. Por ejemplo, está descrito que la formación de mielina está retardada en este trastorno y que hay menor cantidad de cerebrósidos en el sistema nervioso de los enfermos.
A pesar de las consecuencias neurológicas evidentes, es importante iniciar inmediatamente la dieta restringida en fenilalanina para evitar que el daño neurológico progrese, además muchos pacientes, como los casos descritos, presentan regresión de algunos de los síntomas, registrando mejoría clínica evidente del estado neurológico.19-21
Referencias
1. Scriver CR, Kaufman S. The hyperphenylalaninemias. En: Scriver CR, Beaudet A, Sly W, Valle D, editores. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 1667-724. [ Links ]
2. Velázquez A, Loera-Luna A, Aguirre BE, Gamboa S, Vargas H, Robles C. Tamiz neonatal para hipotiroidismo congénito yfenilcetonuna. Salud Publica Mex. 1994; 36: 249-56. [ Links ]
3. Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of new-born infants. Pediatrics. 1963; 32: 338-43. [ Links ]
4. Villasana I, Butler J, Williams JC, Roongta SM. Neurological deterioration in adult phenylketonuria. J Inherit Metab Dis. 1989; 12: 451-7. [ Links ]
5. American Academy of Pediatrics. Newborn screening fact sheets. Pediatrics. 1989; 83: 449-64. [ Links ]
6. Seymour CA, Thomason MJ, Chalmers RA, Addison GM, Bain MD, Cockburn F, et al. Newborn screening for in-born errors of metabolism: a systematic review. Health Technol Assess. 1997; 11: 1-4. [ Links ]
7. Harvey EL, Kirk SF. The use of a low phenylalanine diet in response to the challenging behavior of a man with untreated phenylketonuria and profound learning disabilities. J Intellect Disabil Res. 1995; 39: 520-6. [ Links ]
8. Sweetman L. Newborn screening by tándem mass spectrometry. Clin Chem. 1996; 42: 345-6. [ Links ]
9. Williams K. Benefits of normalizing plasma phenylalanine: impact on behavior and health. A case report. J Inherit Metab Dis. 1998; 21: 785-90. [ Links ]
10. Brown MCJ, Guest JF. Economic impact of feeding a phenylalanine-restricted diet for adults with previously untreated phenylketonuna. J Intellect Disabil Res. 1999; 43: 30-7. [ Links ]
11. Baumeister A. Dietary treatment of destructive behavior associated with hyperphenylalaninemia. Clin Neuropharmacol. 1998; 21: 18-27. [ Links ]
12. Acosta P. Nutrition support of infants, children, and adults with phenylketonuria (PKU). Nutrition Support Protocols (A6224). Ohia, USA: Editorial Ross Laboratories; 2001. p. 1-30. [ Links ]
13. Jefferson JW. Mood stabilizers: a review. En: Dunner DL, editor. Current psychiatric therapy. Philadelphia: WB Saunders; 1993. p. 245-54. [ Links ]
14. Norma Oficial Mexicana para la Atención de la Mujer durante el Embarazo, Parto y Puerperio y al Recién Nacido. NOM-007-SSA2-1993. [ Links ]
15. McCombe PA, McLaughlin DB, Chalk JB, Brown NN, McGill JJ, Pender MP. Spasticity and white matter disease in adult phenylketonuria. J Neurol Neurosurg Psychiatry. 1992; 55: 359-61. [ Links ]
16. Yule KS. Phenylketonuria. En: Ludder JP, Vessey JA, editores. Primary care of the child with a chronic condition. St. Louis: Mosby; 1996. p. 623-49. [ Links ]
17. Koch R, Azen C, Friedman EG, FishlerK, Baumann-Frischling C, Lin T. Care of the adult with phenylketonuria. Eur J Pediatr. 1996; 155 Suppl 1: S90-2. [ Links ]
18. Chase D, Millington DS, Terada N, Kahler SG, Roe CR, Hofman LF. Rapid diagnosis of phenylketonuria by quantitative analysis for phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin Chem. 1993; 39: 66-71. [ Links ]
19. Potocnik U, Widhalm K. Longtermfollow-up of children with classical phenylketonuria after diet discontinuation: a review. J Am Coll Nutr. 1994; 13: 232-6. [ Links ]
20. Yannicelli S, Ryan A. Improvements in behavior and physical manifestations in previously untreated adults with phenylketonuria using a phenylalanine-restricted diet: a national survey. J Inherit Metab Dis. 1995; 18: 131-4. [ Links ]
21. Louis J, Elsas LJ, Acosta P. Apoyo nutricional en la enfermedad metabólica hereditaria. En: Shils ME, Olson JA, editores. Nutrición en salud y enfermedad. Philadelphia: McGrawHill; 2002. p. 1151-214. [ Links ]

