











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
In 1950, human genome mutations were thought to be extremely rare events. A couple of decades later, it was identified that RNA could be retro-transcribed into DNA. That information was to change our way of thinking: our genome was not stable but dynamic. In the following decades, several researchers identified a genome with great variability.1 The discovery of various mutations (DNA alterations with a frequency < 1 % in the general population) or polymorphisms (common genetic variants with a frequency ≥ 1 % in the general population), such as variable number tandem repeats (VNTRs), short tandem repeats (STRs), single nucleotide polymorphisms (SNPs), insertions/deletions (INDELs) and copy number variants (CNVs) has served for identifying regions (or linked genes) or genes associated with various diseases (Table 1).2-7
Table 1 Characteristics of Mendelian and multifactorial diseases (for the latter, only traits of unrelated case-control studies are mentioned)
| Traits | Mendelian disease | Multifactorial disease |
|---|---|---|
| Objective | Identification of the main gene or genes linked in the families | Identification of the gene/genes of small effect size associated with a population base |
| Genetic alteration | Mutation | Polymorphism |
| Inheritance Model | Well defined Autosomal recessive, autosomal dominant, X-linked, uniparental dysomia, etc. |
Undefined |
| Effect of the alteration | High Generally, all individuals who have the mutation in a single gene also have the disease (except for incomplete penetrance) |
Low In individuals who have all associated variants they do not trigger the disease, but cause susceptibility to diseases |
| Frequency of the alteration | Rare Lower than 1% in a population |
Low-common With a frequency of at least 1% in the general population |
| Variants used in these studies | VNTRs, STRs, SNPs | VNTRs, STRs, SNPs, CNVs, INDELs |
| Effect of the environment | Practically none | Strongly influenced |
| Clinical presentation | They occur at the earliest stage of life or during childhood | They frequently occur more towards the stage of young adulthood or adulthood. Others, such as obesity and type 2 diabetes, currently also occur in children |
Genetic variants used in linkage and association studies
VNTRs and STRs have been used in linkage studies to detect regions involved with Mendelian (or monogenic) diseases, such as Duchenne or Becker muscular dystrophy, cystic fibrosis, sickle cell anemia, etc., which due to their low prevalence are quite rare in populations. These diseases have the characteristic that a single altered gene results in a phenotype (Fig. 1), given that the mutation has great biological effect; however, there are always exceptions and, in some cases, even if a mutation occurs, it will not lead to the disease but to incomplete penetrance, i.e., less than 100 % of individuals have the expected phenotype according to their genotype (Table 1 and Figure 1).
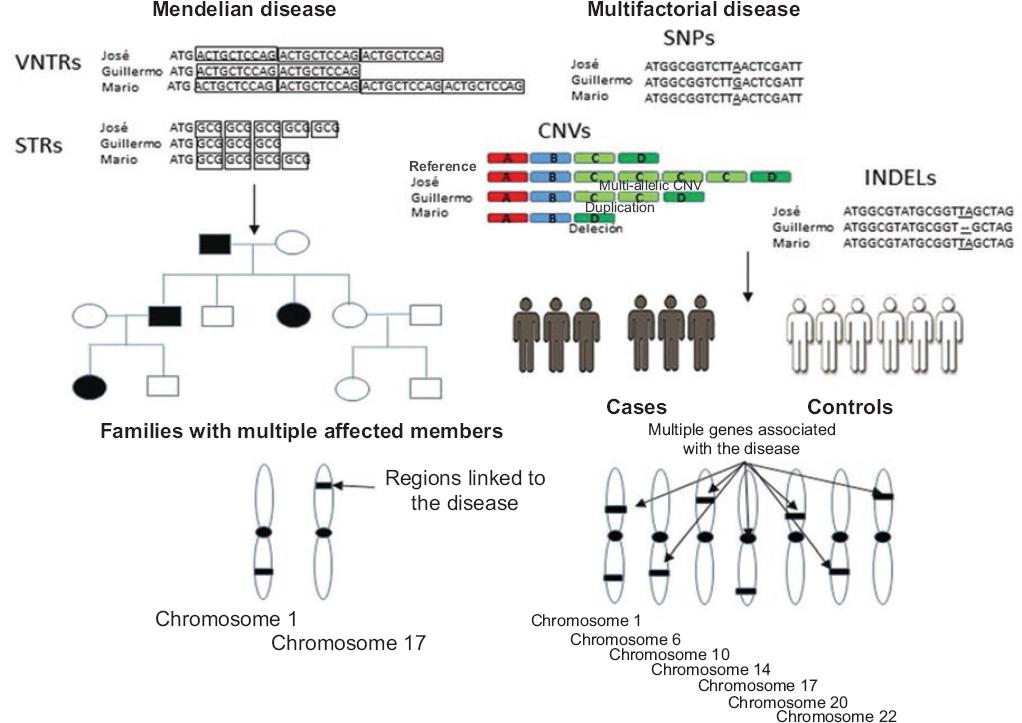
Figure 1 Identification of regions or genes linked by VNTRs, STRs, SNPs, CNVs and INDELs in families or associated genes in unrelated individuals, in different diseases or traits of interest. VNTRs and STRs have been used to map chromosomal regions linked to various Mendelian diseases, where a single or a few genes with great penetrance are responsible for the observed phenotypes. In genetic association studies, VNTRs and STRs were also used to identify genes with small effect size; however, due to their low distribution within the genome and genes, the most commonly used markers in these studies are SNPs, although CNVs and INDELs are also employed.
Linkage (parametric) refers to a co-segregation of these markers with a disease or phenotype in families with multiple affected members; it is measured in a base 10 logarithm (LOD score) and it must show at least a LOD score of 3, which means that the genetic marker shows a 1000-fold linkage with the trait of interest. Thus, a linkage positive signal (LOD score ≥ 3) serves to identify chromosomal regions (although not necessarily the gene) involved in Mendelian pathologies (Fig. 1).8-10 Since this strategy does not detect the linked gene, but rather a chromosomal region, the next step is to detect the mutation in a nearby gene involved with the disease by other methodologies (Fig. 1).
On the other hand, in association studies, where the co-occurrence of a genetic marker with a disease or phenotype of interest is observed,8,9 VNTRs, STRs, SNPs, CNVs and INDELs are used with the purpose to detect genes associated with the trait of interest (Fig. 1). Given their abundance in the genome and relative ease to assess them with automated methods, SNPs are routinely used in this type of analysis.6,7 Their use in case-control investigations is highly convenient (this article only refers to case-control studies), since a much finer genetic mapping can be carried out versus VNTRs or STRs, which are scarce if compared to SNPs. Some SNPs can be assessed using studies of candidate genes (genes that encode or not proteins and that due to their biological effect participate in the pathogenesis of a disease), or thousands of them can be examined by means of genome-wide association studies (GWAS), which detect dozens of loci related to these traits (in these studies, no hypotheses are established because they do not target any particular gene).
It is important noting that, in order to obtain reliable results in GWAS, they must include hundreds or thousands of samples of cases and controls and be replicated in at least a second study group, corrections should be made and the Bonferroni test should be included (if one million SNPs are evaluated, then p-values = 0.05/10-6 should be established, i.e., 5 x 10-8), as well as a correction for ancestry (through informative markers of ancestry), etc.; some functional test of associated variants is also suggested.11 Linkage and association studies have several differences (Table 1).
Mutation or polymorphism
In Mendelian or multifactorial diseases, mutations and polymorphisms, although representing a change in the DNA sequence, are defined differently due to their allelic frequency and biological effect in an individual (Fig. 2). In Mendelian diseases, mutations are mentioned, while in multifactorial diseases, polymorphisms are referred to. In the former, a single mutation in a gene (with a frequency < 1 %, which does not occur in the general population) is enough for cystic fibrosis, Duchenne or Becker muscular dystrophy, among others, to occur (except in case of incomplete penetrance), and practically they are not influenced by the environment (Fig. 2). In these diseases, mutations follow a well-defined heritability pattern and include autosomal recessive, autosomal dominant, X-linked, etc. inheritance.12
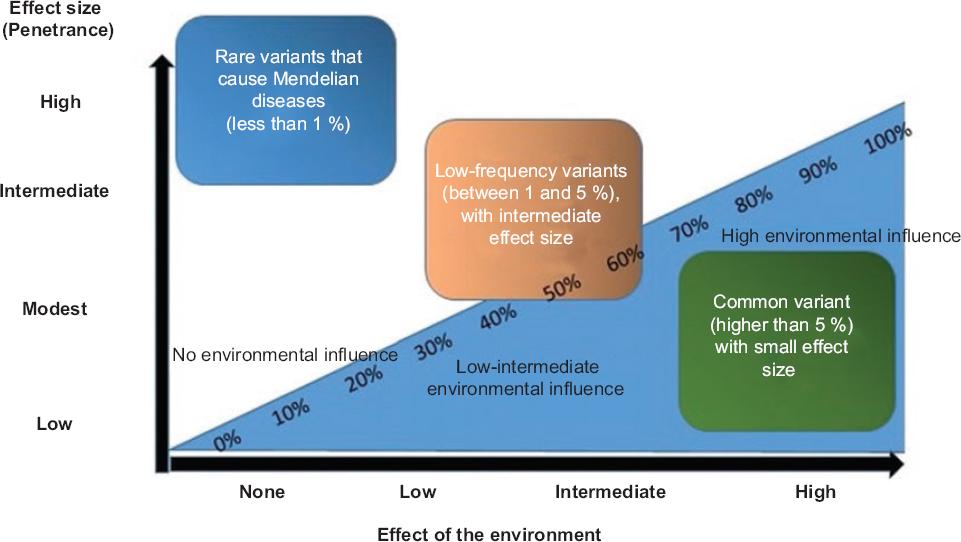
Figure 2 Image depicting the effect of the environment; and of mutations-polymorphisms in the genesis of Mendelian and multifactorial diseases. The environment has little or no effect on the genesis of most Mendelian diseases (caused by mutations); in contrast, it can influence up to nearly 100 % on multifactorial diseases (multiple genes and with a small effect size), while gene variants can have little or much relevance in the development of these diseases.
In turn, multifactorial diseases are influenced by tens or hundreds of (polygenic) susceptibility genes, and each loci contributes with a small effect size (Fig. 2). Additionally, the alleles of the associated variants have frequencies that range from low to normal (Fig. 2), without a well-defined inheritance pattern (owing to this trait, they are also defined as complex diseases), with environmental risk factors being involved in their development; i.e. even with all risk alleles, an individual will not develop the disease unless there is an environmental risk factor that triggers its development (Table 1 and Figure 2);6,7 for example, in some autoimmune diseases such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), more than 100 genes associated with susceptibility have been identified; however, these are not sufficient to trigger the disease.13,14
VNTRs in Mendelian and multifactorial diseases
VNTRs, also known as mini-satellites, comprise tandem repeats of non-coding sequences of 10 to 100 nucleotides in length (Fig. 1). Approximately 1500 VNTRs have been calculated to exist in the human genome.1,4,10 Their use as markers for linkage studies occurred in the early 1980s, when they were used to map chromosomal regions linked to various Mendelian diseases (Fig. 1). VNTRs were useful for identifying signals close to the main causative genes of these diseases; subsequent studies identified those genes.8,10
On the other hand, the use of VNTRs in linkage analyses for complex diseases were not as successful as expected, since these diseases are influenced by several genes with a small effect size (there is not a main gene).8 In addition, VNTRs have been used in genetic association studies, although currently they have been practically forgotten mainly due to their low number and because the vast majority are located at chromosomal terminal regions. However, not studying them potentially implies losing (lossing heritability) some loci involved in this type of diseases. A 28-bp VNTRs located in the HRAS oncogene has been proposed to cause susceptibility to several types of cancer through the following mechanisms:
Linkage disequilibrium, i.e., some alleles of this VNTRs are in linkage disequilibrium with the true variant associated in those tumors.
Disruption of rel-NF-kB transcription factors gene expression, since it is located at a binding site for that family of transcription factors and could be a direct cause of the association.1
On the other hand, a VNTRs with four repeated 27 bp was recently identified, which is located in exon 4 of the endothelial nitric oxide synthase (eNOS3) gene that confers susceptibility to coronary artery disease. Correlation studies showed that this VNTRs is associated with high serum triglyceride levels (versus that of five 27 bp repeats), while the in silico analysis showed that this repeat can affect alternative splicing.15
Other VNTRs located in myoglobin, interleukin-1 receptor antagonist, DNase 1, and interleukin-4 genes, among others, have been observed to be associated with RA, SLE and osteoarthritis (Table 2).16-18
Table 2 Genetic variants involved in Mendelian and multifactorial diseases
| Variant | Mendelian disease | Multifactorial disease | References |
|---|---|---|---|
| VNTRs | Hypertension | Various types of cancer, rheumatoid arthritis, SLE, osteoarthritis | 1,15-18 |
| Linked chromosomal regions: 2p22.1, 5q33 | Genes: HRAS, IL1, IL4, DNase 1 | ||
| STRs | Different STRs have been linked in more than 40 Mendelian diseases; these diseases include spinocerebellar ataxia, Huntingtons disease, spinobulbar muscular atrophy, myotonic dystrophy | Various types of cancer, autoimmune diseases | 3,19-30 |
| Hungtintin gene, ATXN3, DM1 | Genes: KCNQ1OT1, PTPN11, ECRG2, TCR | ||
| SNPs | | Various types of autoimmune, metabolic, etc., diseases. | 6,7,38,39 |
| Genes: PTPN22, TNF, FCRL3, microRNA, FTO, LEPR, etc. | |||
| CNVs | Autosomal dominant microtia, neurodevelopmental diseases such as autism and epilepsy, congenital heart defects, etc. | Various cancers, autoimmune, mental diseases, etc. | 40-43 |
| Identified chromosomal regions: 4p16 | Genes: FCGR3B, C4, CCL3L1, NBPF, UGT2B17 | ||
| INDELs | | Multiple sclerosis, SLE, obesity and body mass index | 46,48 |
| Genes: BAFF, LEPR, UCP2 |
VNTRs = variable number tandem repeats, STRs = short tandem repeats, SNPs = single nucleotide polymorphisms, CNVs = copy number variants, INDELs = insertions/deletions, SLE = systemic lupus erythematosus, HRAS = proto-oncogene, IL1 = interleukin 1, IL4 = interleukin 4, ATXN3 = ataxin 3, KCNQ1OT1 = KCNQ1 overlapping transcript 1, PTPN11 = protein tyrosine phosphatase non-receptor type 11, ECRG2 = esophageal cancer-related gene 2, TCR = T-cell receptor, PTPN22 = protein tyrosine phosphatase non-receptor type 22, TNF = tumor necrosis factor, FCRL3 = Fc receptor-like protein 3, FTO = obesity and lean mass-associated protein, LEPR = leptin receptor, FCR3B = Fc fragment of IgG low affinity IIIb receptor 3, C4 = complement protein 4, CCL3L1 = C-C motif chemokine ligand 3-like 1, NBPF23 = NBPF pseudogene, UGT2B17 = UDP glucuronosyltransferase family 2, member B17; BAFF = B cell activating factor, UCP2 = uncoupling protein 2.
STRs in Mendelian and multifactorial diseases
STRs (also known as microsatellites) are tandem-repeats that range from 2 to 10 bp, although some authors define them as 2 bp-repeats (Fig. 1). Due to their wide distribution in the human genome, they reach up to 3 % of total nucleotides, accounting for nearly 6 % of coding regions (where the 5 untranslated, coding sequence and 3 UTR regions are included).3 Recently, a first catalog of human STRs has been reported at the level of wide population-based genome studies: nearly 700,000 STRs have been identified in 1009 individuals in phase 1 of the 1000 Genomes Project, an outstanding finding because it will contribute to identify part of the missing heritability in multifactorial diseases.3
STRs are found in different parts of the structure of protein-coding genes and can have an effect on gene expression, RNA stability and creation or destruction of an open reading frame, etc.3 They have high hypermutability, i.e., the repeats tend to generate more copies due to recombination or replication errors. These variants have been used as genetic biomarkers in linkage studies with more success than VNTRs due to their wider distribution.8 For example, some studies in RA (a complex and multifactorial disease) showed that several STRs localized in chromosome 6p21 are important for this pathology; simultaneously, other linked regions in this autoimmune disease were identified with these markers.19,20
On the other hand, several STRs have been linked to more than 40 Mendelian diseases, including spinocerebellar ataxia, Huntingtons disease, spinobulbar muscular atrophy, among others, which originate from a dramatic expansion of trinucleotides and behave as dominant gain-of-function mutations.3,21
The CAG STRs (a codon involved in glutamine encoding), located in the coding sequence of the huntingtin gene, has been identified as the responsible for Huntingtons disease (Table 2); in 1993, less than 35 repeats were identified in healthy individuals, while those affected had 37 or more glutamine repeats, which leads to a change in the structure and function of the protein.22,23 Another example is spinocerebellar ataxia type 3 (Table 2), in which an expansion of the CAG trinucleotide in the coding sequence of the ATXN3 gene (which encodes the ataxin 3 protein) is also associated with abnormal folding of the protein and with an accumulation in certain regions of the brain.24 On the other hand, the CTG repeat, located in the gene that encodes protein kinase MD1 has been implicated in myotonic dystrophy type 1 and the GGGGCC repeat of the C9ORF72 hypothetical gene is involved in the onset of frontotemporal dementia.25
Regarding the role of STRs in multifactorial diseases, several of them have been associated with susceptibility; for example, the STRs located in the KCNQ1OT1 gene (long non-coding RNA gene involved in the silencing of a cluster of genes in cis) causes a decrease in the risk for the development of hepatocellular carcinoma.26 Other STRs located in genes such as PTPN11, ECRG2 and TCR have been associated with the development of various cancers and autoimmune diseases (Table 2).27-30
SNPs and multifactorial diseases
SNPs are single-nucleotide variants, generally biallelic, and represent the most abundant polymorphisms of human genome (found every 200 to 500 bp). Recent data from 1000 Genomes Project indicate that there are nearly 11 million of them and each individual has an average of three million.5,31,32 Given their wide distribution, SNPs are found in regions where there are genes present or not. In the former, they are located in any part of its structure and include promoters, exons and introns, while SNPs located in non-coding RNA genes can be found in the promoters or in the non-coding sequence.7 From a functional point of view, SNPs located in protein coding and non-coding genes affect various processes that are mentioned in detail in the article Functional implications of single nucleotide polymorphisms (SNPs) in protein-coding and non-coding RNA genes in multifactorial diseases.7
Since their discovery, SNPs have been used in association studies.7,33 Associations between these variants and multifactorial traits are measured according to their statistical significance, which is generally p = 0.05 for candidate gene studies or p = 5 x 10-8 for GWAS.11,34 However, it is important taking into account certain deficiencies in candidate gene investigations, including population stratification, low sample size, low statistical power, etc.,9,11,33 which do not represent a problem in GWAS, whose analysis includes evaluating various ancestry markers, a large sample size (hundreds or thousands of case-controls), replications of the findings in other samples, as well as elimination of other biases that may confound associations.11 Through both strategies, multiple genes associated with multifactorial diseases have been recognized;6,7,11,33 for example, a candidate gene study identified that SNPs in pre-miRNAs 146a and 499 confers susceptibility to SLE,35 while the non-synonymous C1858T SNPs which leads to a change of arginine for tryptophan at position 620 of the PTPN22 mRNA confers susceptibility to RA36 and Graves disease,37 since it affects the structure and function of the protein, as well as various immune processes.36 The C allele of FCRL3 SNPs-169T/C which codes for Fc receptor-like protein 3 modifies binding affinity for NF-kB transcription factor and is correlated with higher levels of mRNA of this receptor involved in B-cell activation, which causes susceptibility to SLE and RA (Table 2).6,7
On the other hand, a recent analysis of 225 SNPs recorded in a database of polymorphisms previously associated with obesity, identified that 15 variants of the FTO gene are associated with body mass index, and thus it replicated previous findings and confirmed that FTO represents the main risk locus for this trait.38 Although these variants are found in intron 1 of this gene, functional studies show that some SNPs can form long-range connections to regulate the expression of the IRX3 gene, which has been implicated in some obesity-related events through FTO.38 Another study has shown the role of various SNPs located in the leptin receptor gene (LEPR) and their relationship with susceptibility to obesity.39 There are other examples about the role of SNPs and their relationship with multifactorial diseases.6,7
CNVs in Mendelian and multifactorial diseases
CNVs (a type of structural variation) encompass segments of DNA with a length equal to or larger than 1 kb, which occur in a variable number of copies between individuals and comprise deletions, duplications and insertions (Fig. 1).40 CNVs have been suggested to influence on up to 1 % of variability between two individuals, while SNPs account for about 0.1 %.40 In addition, CNVs can cause Mendelian diseases and susceptibility to multifactorial diseases.40-42 Unfortunately, the amount of CNVs identified in the human being is not defined, since it depends on the nature of the arrangement used to assess them. Currently, in the CNVs database, there are 29,133 included, with 41 % overlapping with known genes, which means that an important part of them can cause Mendelian or multifactorial diseases.43 From the biological-pathological effect point of view, CNVs are the most important variants (due to their size) related to susceptibility for various human diseases (when compared to VNTRs, STRs and SNPs). They have been reported to cause Mendelian diseases in neonatal or early stages of childhood, including neurodevelopmental diseases such as autism or epilepsy and congenital heart defects, among others (Table 2).43
Regarding multifactorial diseases, an 8-kb CNVs localized in the NEGR1 gene has been recently shown to be associated with protection against obesity, and a functional study indicated that the NKX6 transcription factor binds to this region and represses its transcription, and thus the deletion of this CNVs prevents the binding of this transcription factor. Additional tests are still required to better understand its role in obesity.39
On the other hand, several CNVs located in FCGR3B, C4, CCL3L1, and DEBF have been associated with the development of glomerulonephritis in patients with SLE, HIV/AIDS and psoriasis, respectively (Table 2).2,41-43 It is important mentioning that the data on genetic association of CNVs located in FCGR3B, CCL3L1, DEBF and C4 should be taken with great caution since these associations should be identified with SNPs microarray studies; however, so far there is no evidence of association of these variants using these molecular tools.
INDELs and multifactorial diseases
Recently, small INDELs (Fig. 1) have been reported in the human genome, which comprise a length of 1 to 10,000 bp.44 A study on chromosome 22 showed that out of 100 % of variants, 13 % corresponded to INDELs.44 Other research carried out on 330 genes from different individuals reported 2,393 small INDELs from 1 to 543 bp in length.44 On the other hand, 3.4 million of small INDELs were identified, out of which 1.96 million were non-redundant, while 819,363 were located in human genes, out of which 2,123 were found in exons. The final number of INDELs identified in the genome is also not established, since it depends on the last-generation tool used and the amount of INDELs each person has; for example, the Watson genome contains 222,718, while the Venter genome possesses 823,396.44
Given their recent identification, INDELs have not been assessed in linkage analyses, but in association analyses they have. From the functional point of view, and given that small INDELs (especially those that cover some tenths of bp) can be found in genes and affect promoters, exons (5 UTR, coding sequence, and 3 UTR) and introns, and possibly influence on the development of multifactorial diseases because they are also capable of altering aspects of mRNAs or proteins, for example, 184 INDELs out of 2,123 are close to the binding sites between exons and introns, thus affecting splicing and mRNA-proteins that are useful for cells; the remaining 1939 are located in the coding sequence and cause alterations in the reading frameshift or in premature termination of the protein.45,46
Regarding the role of these variants and their genetic association with some diseases, the GCTGT-> A INDELs (the A allele confers risk), located in the 3 UTR region of the TNFSF13B gene (which codes for the B-cell activating factor cytokine) has been reported to confer risk for the development multiple sclerosis and SLE in patients from various European countries (Table 2).47 It is important mentioning that variant A of the GCTGT-> A INDELs generates a short mRNA that escapes the inhibition of a microRNA (a negative regulator of the expression of various mRNAs at the post-transcriptional level), which leads to increased production of the soluble B-cell activating factor cytokine in these patients, thus causing susceptibility to the development of said diseases.47
On the other hand, the CTTTA INDELs localized at the 3 UTR region of the LEPR gene is associated with body weight increase,48 while the 45-bp INDELs located in the 3 UTR region of the UCP2 gene (which codes for an uncoupling protein that acts as a mitochondrial transporter and homeostasis and thermogenesis energy regulator) has been associated with body mass index and body weight alterations after food consumption.48
Another study assessed a group of INDELs in various genes of patients with colorectal cancer; these variants in the ACE, UCP2, TYMS, IL4, NFKB1, CASP8, TP53, HLAG, UGT1A1 and SGSM3 genes were found to be associated with susceptibility and some clinical traits (Table 2).49 Other small INDELs located in the PPP3R1, PARP1, pre-miR3131, COL1A2 and HLA-G genes have shown an association with susceptibility for coronary artery disease, melanoma, hepatocellular carcinoma, osteoporosis and SLE, respectively.50-54
Conclusions
Genetic markers such as VNTRs, STRs, SNPs, CNVs and INDELs have been useful for identifying genes related to Mendelian and multifactorial diseases. Since their appearance, VNTRs and STRs were used in linkage studies to identify regions involved with rare diseases; however, in common diseases they were not successful. With SNPs subsequent identification, they became the genetic markers of choice for genetic association studies. Currently, with the analysis of INDELs and CNVs through various tools of low, medium and high density, we have begun to understand their role in the genesis of Mendelian diseases and in susceptibility to multifactorial diseases. In the not too distant future, knowing the genomic variability involved in both types of diseases will result in the development of individualized, preventive and predictive medicine.

