











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkDiscovering the path of research
In 2009, I delivered a plenary lecture at the 44th Congreso Mexicano de Química that was held in Puebla, Mexico from 26-30 September of 2009. An outgrowth of that meeting was an article published in the Journal of the Mexican Chemical Society that was entitled “People, travel, seminars, reading, the classroom, and curiosity - sources of research inspiration: aspects of organic redox chemistry”. [1]
I enjoyed all aspects of the endeavor - the scientific discussions in Puebla, renewing friendships and making new ones, the social events, and the process of writing the manuscript. Now, I am pleased to pay tribute to Elsa Arce, Joan Genescá, Ignacio González, Jorge Ibañez, Yunni Meas, Omar Solorza, emeritus SNI researchers in the area of electrochemistry, for their many contributions, as well as for their stewardship as ambassadors for the field throughout the world.
The present contribution overviews several aspects of my groups’ involvement in the field of mediated electron transfer. From the laboratories of its pioneers to the present day, the field maintains a position of interest and fundamental importance and is now widely accepted and utilized. I begin with a brief review of the basics where we will:
differentiate mediated and direct electrolysis;
highlight similarities between it and sensitized photolyses;
explore the steps/stages associated with a mediated process focusing particularly upon the kinetics and thermodynamics of the electron transfer step;
describe how to overcome the thermodynamic impasse that frequently accompanies electron transfer.
These ideas will be illustrated using examples from research carried out in my group at UCSB. Both reductive and oxidative processes will be described, and the so-called Zeng and Francke mediators will be introduced and their application to catalysis and redox neutral processes will be illustrated. We will also show that a single structure can be used to initiate either a reduction or an oxidation simply by choosing whether the active state is generated photochemically or electrochemically. There are undoubtedly many more examples. What are they? Where can one look to find them? The territory is fertile and awaits rewarding exploration.
Mediated electron transfer
Unlike a direct electrolysis, an indirect electrolysis occurs using a redox mediator, a substance that intervenes between the electrode and the substrate. [2] The figure shown below illustrates the anodic oxidation of the mediator, M, to convert it to its active form M+•. Like a sensitized photolysis wherein the input energy is matched to that of the sensitizer and not the substrate, the potential is set to match that of the mediator rather than the substrate. Once formed, M+• can engage in a homogeneous electron transfer with a substrate, Sub, to form the desired reactive intermediate, Sub+•, and return the mediator to its resting state, M. In this way M+• undergoes turnover and is returned to its initial form, M, leading to a so-called catalytic current that can be witnessed by an increase in current. [3]
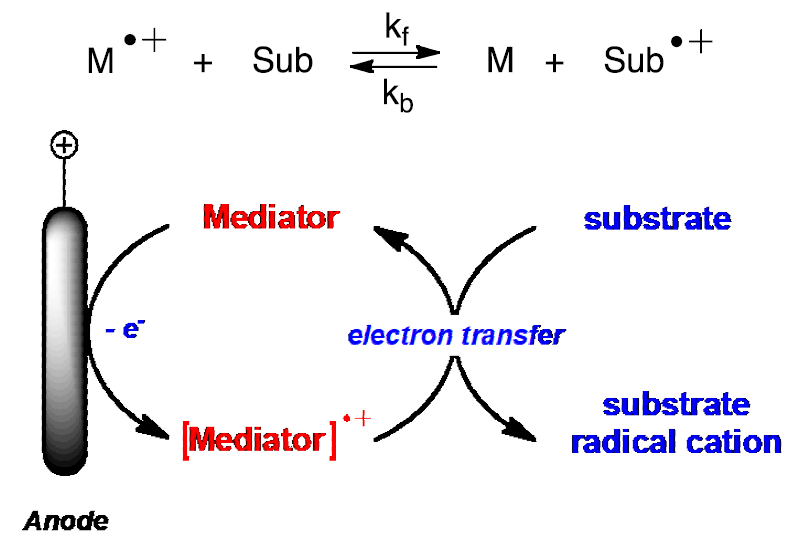
Turnover and return of the mediator cation radical to its resting state; this leads to a "catalytic current" (top). Homogeneous electron transfer to a substrate (bottom).
Care should be exercised to ensure that M+• is not prematurely returned to the resting state by migrating to the cathode. In an effort to limit migration, one can increase the distance between electrodes, an option that can easily be implemented by using a two-compartment cell.
Electroreductive cyclization via both direct and indirect (mediated) electrolysis
The so-called electroreductive cyclization (ERC) reaction is initiated via cathodic reduction of an electron deficient alkene that is tethered to an acceptor, generally an aldehyde or a ketone functional group. [4] The reaction leads to the formation of a new sigma bond between two formally electrophilic centers, thus providing an example of umpolung reactivity. [5] Detailed investigations established that the rate determining step is protonation of the initially formed radical anion. [6] The ERC reaction shown below was used as a key step of a formal total synthesis of the antitumor antibiotic called quadrone. [7] A noteworthy feature is the fact that cyclization occurred in a sterically crowded environment that led to formation of the quaternary center found in the natural product.
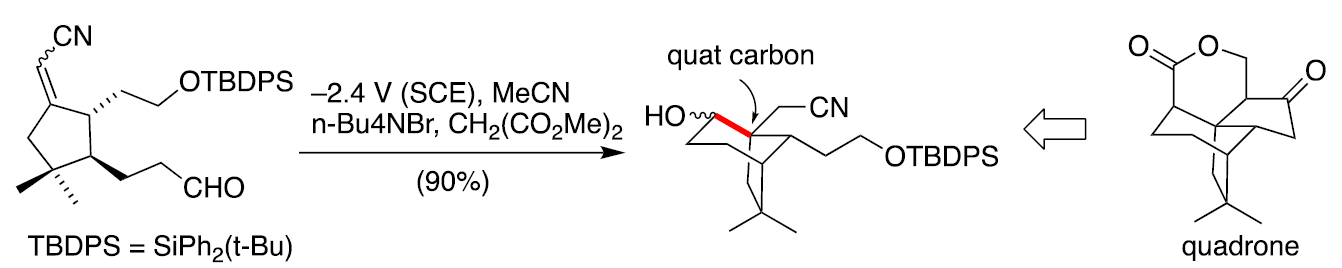
Reaction 1 Use of the electroreductive cyclization reaction en route to the antitumor agent called quadrone.
ERC reactions can also be conducted in an indirect manner using nickel salen as a redox mediator. [8] For the controlled potential electrolysis portrayed below, the potential was set at -2.1 V (Ag/AgNO3), a value corresponding to the peak potential for reduction of nickel salen, rather than -2.7 V (Ag/AgNO3), the peak potential corresponding to reduction of the substrate. Thus, one is able to operate at a potential that is 600 mV shy of that that would be used in a direct electrolysis, thereby that would lead to a thermodynamically unfavorable electron transfer step.
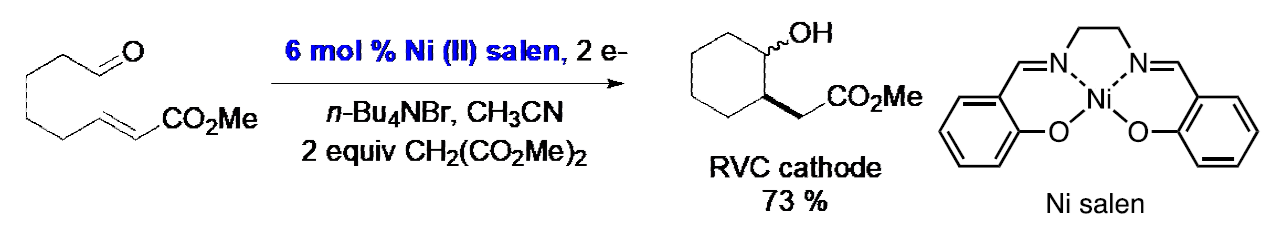
The unfavorable electron transfer equilibrium constitutes a “thermodynamic bottleneck” and is a common occurrence in mediated processes. Fortunately, the equilibrium can be shifted toward the product provided there exist one or more rapid followup reactions to drain the equilibrium toward the adduct.

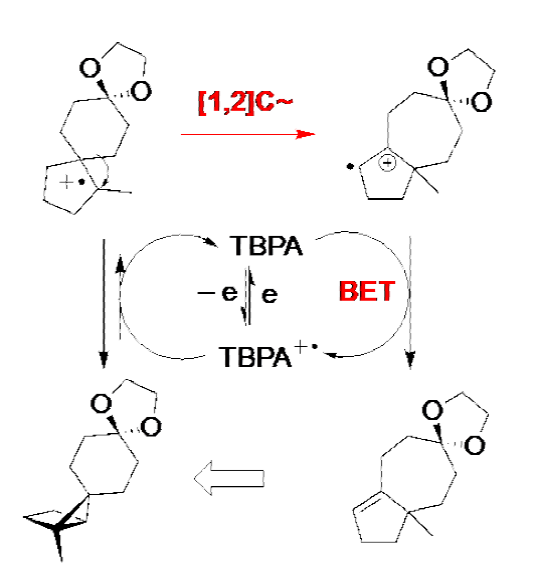
Use of a triarylamine mediator to achieve a total synthesis. Driving unfavorable equilibria in a redox neutral setting. A cautionary tale
Tris(4-bromophenyl)amine (TBPA) is undoubtedly one of the most widely used mediators. [9] Park and Little used it to mediate the redox neutral rearrangement of the highly strained structure portrayed in the equation shown below, in a key step ultimately leading to the synthesis of the simple sesquiterpene called daucene [10] In the present instance, a total of 40 mol % of TBPA was used. This ridiculously large quantity could have been avoided had an H-cell been used. The vial (i.e., a single compartment cell) that was used placed the cathode close enough to the anode so that the active form of the mediator, TBPA+•, was given a choice between oxidizing the substrate and migrating to the nearby cathode where it could undergo reduction to reform TBPA in an energy wasting step.
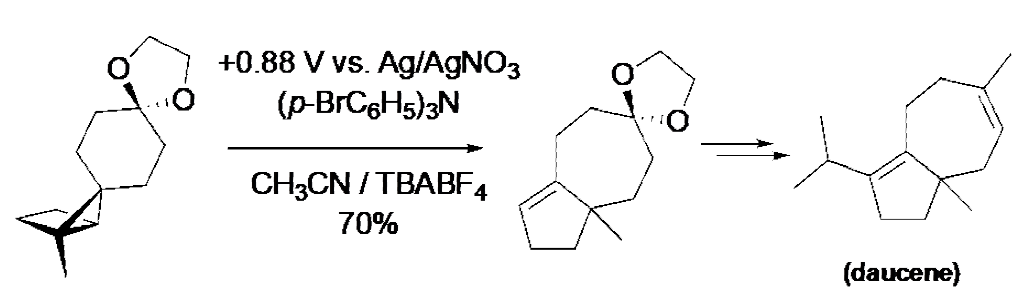
Reaction 3 Use of TBPA to mediate a key step in a total synthesis of the sesquiterpene called daucene.
Like the ERC chemistry previously described, an unfavorable electron transfer equilibrium occurred, this time as a consequence of the fact that the potential was set at that of TBPA (+ 0.9 V, Ag/AgNO 3) rather than that of the substrate (+1.4 V, Ag/AgNO3). Once again, the key to success was the existence of one or more followup reactions that served to drain the equilibrium toward the product. In the present instance, both the exothermic (1,2)-carbon migration (a Wagner-Meerwein rearrangement), and the back-electron transfer (BET) step shown below, are likely candidates in this overall redox neutral transformation.
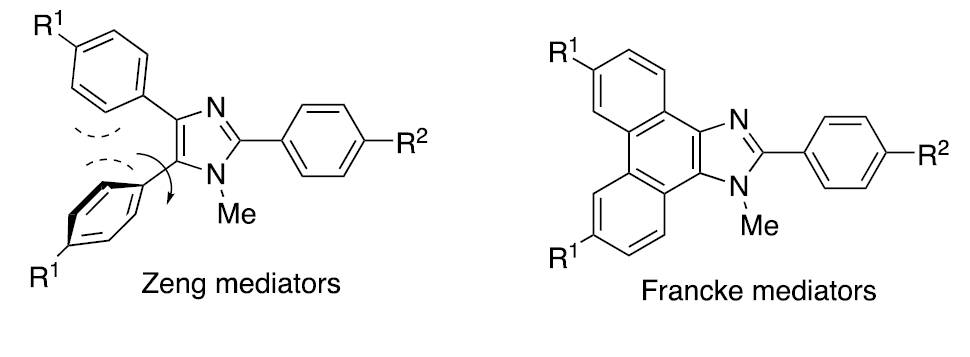
Triarylimidazole aka “Zeng mediators”
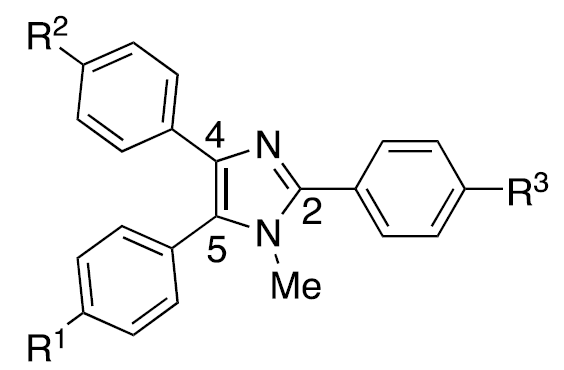
In addition to the multitude of triarylamine mediators that exist, a new class of mediators became available as a result of a collaboration with the Zeng group in Beijing. [11] These so- called “Zeng mediators” are based upon a triarylimidazole framework. Variation of the electronic character of the para-substituents appended to the aromatic rings allows access to mediators whose oxidation potentials span a range of >700 mV. Of the more than 30 structures that have been synthesized, one where the substituents are R1 = R2 = H, R3 = Br has been used most often. For convenience, we refer to this structure as HHBr, an abbreviation that reflects the substituents.
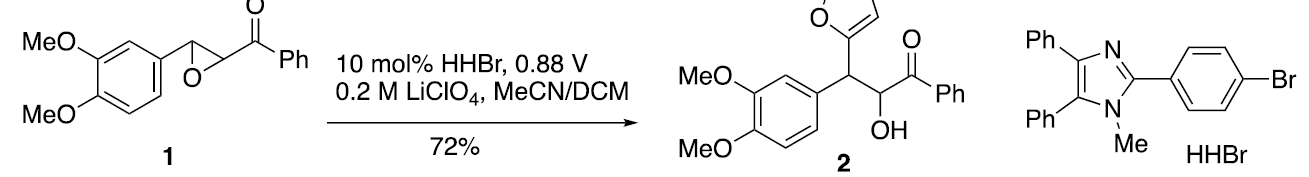
A striking example of the use of HHBr is the Friedel-Crafts-like β-heteroarylation of chalcone epoxides as shown below. 12] Here, the active form of the mediator is generated by oxidizing HHBr at +0.88 V (Ag/AgNO3), a value that is 160 mV less than that required for direct oxidation of the substrate (+1.06 V, Ag/AgNO3). Like the nickel salen and TBPA mediated processes discussed earlier, the present case also requires the existence of one or more followup reactions to drain a thermodynamically unfavorable electron transfer equilibrium. Once the starting material has been oxidized, the sequence proceeds in the manner portrayed below. Thus, ring opening of the epoxide reveals a β-aryl stabilized cation 4 that is subsequently intercepted by a heterocycle, in this case, furan, to form structure 5. Subsequent H-atom transfer (HAT) leads to 6, a structure that formally corresponds to a C-2 substituted cation radical of furan. Since furan oxidizes at ~1.5 V, 6 is a powerful oxidant. Being generated in situ, electricity is no longer required to keep the sequence going.
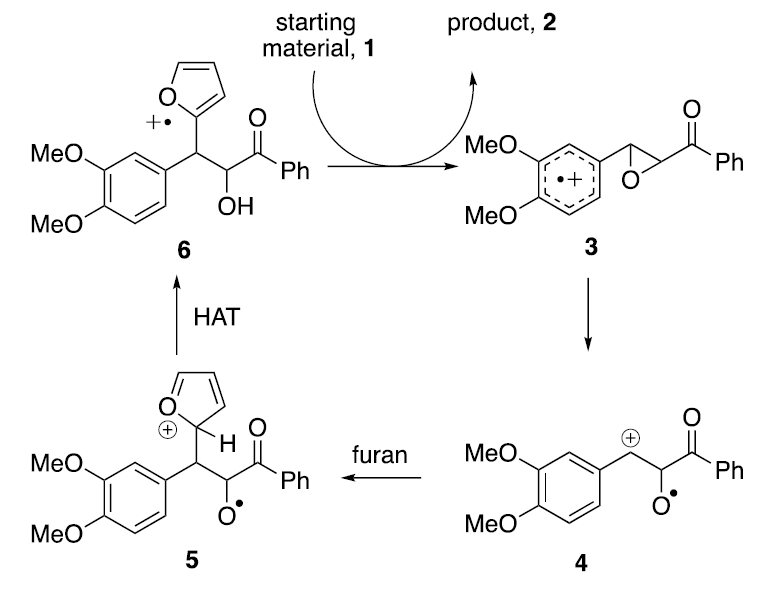
As the sequence proceeds, the powerful oxidizing agent, structure 6, is formed in situ, thereby setting up a chain process.
Improved properties - the “Francke mediators”
Just as the electronic character of the substituents appended to the aryl rings of triarylamines influence the range of accessible potentials, so too is the case for the Zeng mediators. Unfortunately, the aryl groups positioned at C4 and C5 can undergo a propellor twist that precludes the full expression of that character via resonance and induction. Fortunately, this is easy to remedy simply by flattening the framework into that of a phenanthroimidazole to afford the so-called Francke mediators. [13]
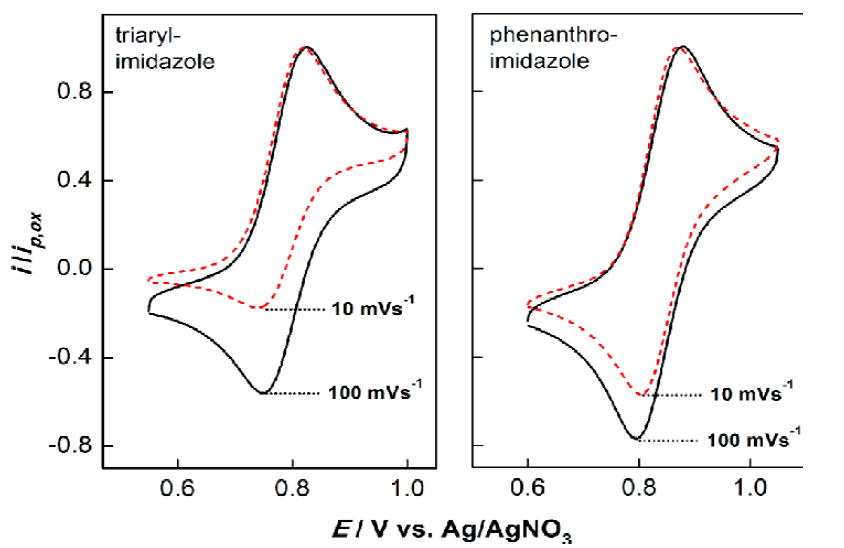
Indeed, flattening does enhance the role of the substituents. For example, for the compounds synthesized at the time of our original publication in 2014, the range of potentials increased by approximately 40 % compared to triarylimidazoles bearing the same substituents. In addition, the cation radicals displayed increased stability as is evidenced by comparing cyclic voltammograms for the two frameworks shown in Fig. 1 as a function of scan rate. On the left is the voltammogram for the Zeng mediator where R1 = H and R2 = OMe; on the right, we see the voltammogram for the Francke mediator bearing the same substituents. Notice the substantial decrease in the size of the reduction current for the triarylimidazole compared with its phenanthroimidazole counterpart when the scan rate is slowed from 100 to 10 mV s-1.
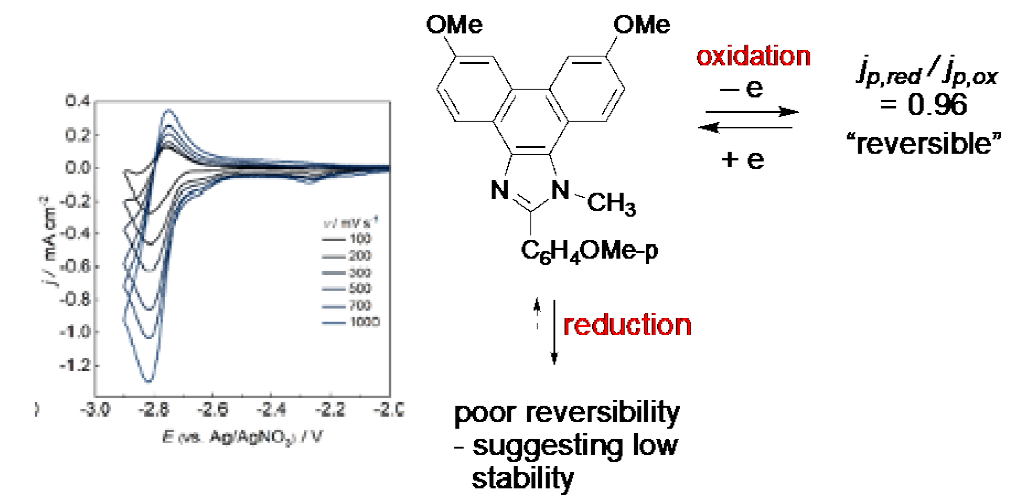
In principle, the active form of the Francke mediators could serve either as oxidizing or reducing agents. In practice, they are oxidizing agents. This preference is easy to understand when one compares the cyclic voltammograms for oxidation and reduction of the Francke mediator where R1 = OMe and R2 = C6H4OMe-p, it serving to illustrate the behaviour of the class. Oxidation curves (not shown) are characterized by nearly fully reversible behaviour. This is in stark contrast with reduction where, as shown, the radical anion decays rapidly rendering it useless as a potential mediator.
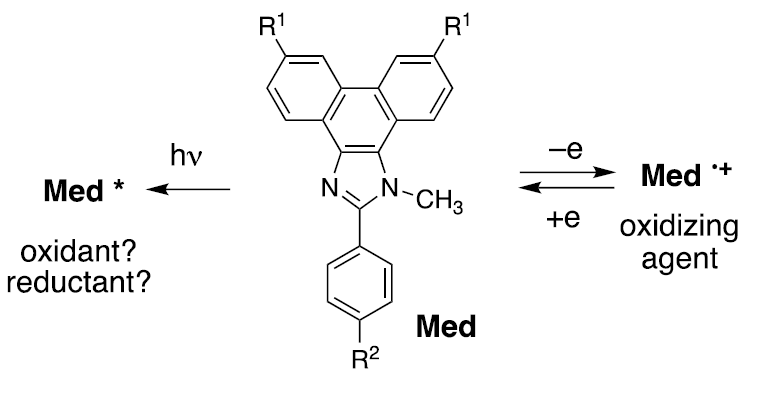
Transitioning to ‘duality’
Prof. Dr. D. Hollmann of LIKAT measured the ESR spectrum for the cation radical derived from the unsubstituted Francke mediator 1-methyl-2-phenyl-1 H-phenanthro[9,10-d]imidazole (R1 = R2 = H), and determined the halflife for decay of the signal to be a remarkably long 44 s. Once again, the stability of the Francke mediator-derived cation radicals is evident.
Having been trained as a photochemist, it was natural to wonder what the properties of the excited state might be (see Fig. 3). In principle the excited state could serve as an oxidizing agent toward a substrate, thereby converting the excited state to a radical anion. Alternatively, it could serve as a reducing agent and be converted to the same cation radical that is encountered when the mediator is used to mediate electrooxidation. Intuitively, given the remarkable stability of the cation radicals, the latter seems like the more reasonable alternative. Should this prove accurate, then a rather remarkable outcome would result. That is, depending upon whether light or electricity was used to initiate the chemistry, it should be possible to use a single structure to mediate either an oxidation or a reduction, at will and by choice.
To probe these ideas and explore the photochemistry, eight phenanthroimidazoles whose structures varied as a function of the substituents, were irradiated using a bank of three 405 nm LEDs. [14] Four aryl halides, Ar-X, were chosen as substrates (S), and n- Bu3N was selected as a sacrificial electron donor. Let’s focus upon the excited state behavior of the Francke mediator labeled T (R1 = R2 = OMe), the letter standing for test case. A Rehm-Weller-like expression was used to estimate the Gibbs free energy (ΔG 1 ) of electron transfer between T* and the potential reaction partners. [15] In the equation shown below, 0,ET0refers to the energy for the 0,0 transition of the emission spectrum for T*. Using experimental values, we determined that for an oxidative quench that converts T* to T •+ , ΔG ET < 0, and is, therefore, thermodynamically favorable. In contrast, for a reductive quench converting T* to T •-, ΔGET > 0, and is therefore predicted to be thermodynamically unfavorable. These outcomes accord with the low stability of the Francke mediator radical anions, and predict that T* should serve as a reducing agent.
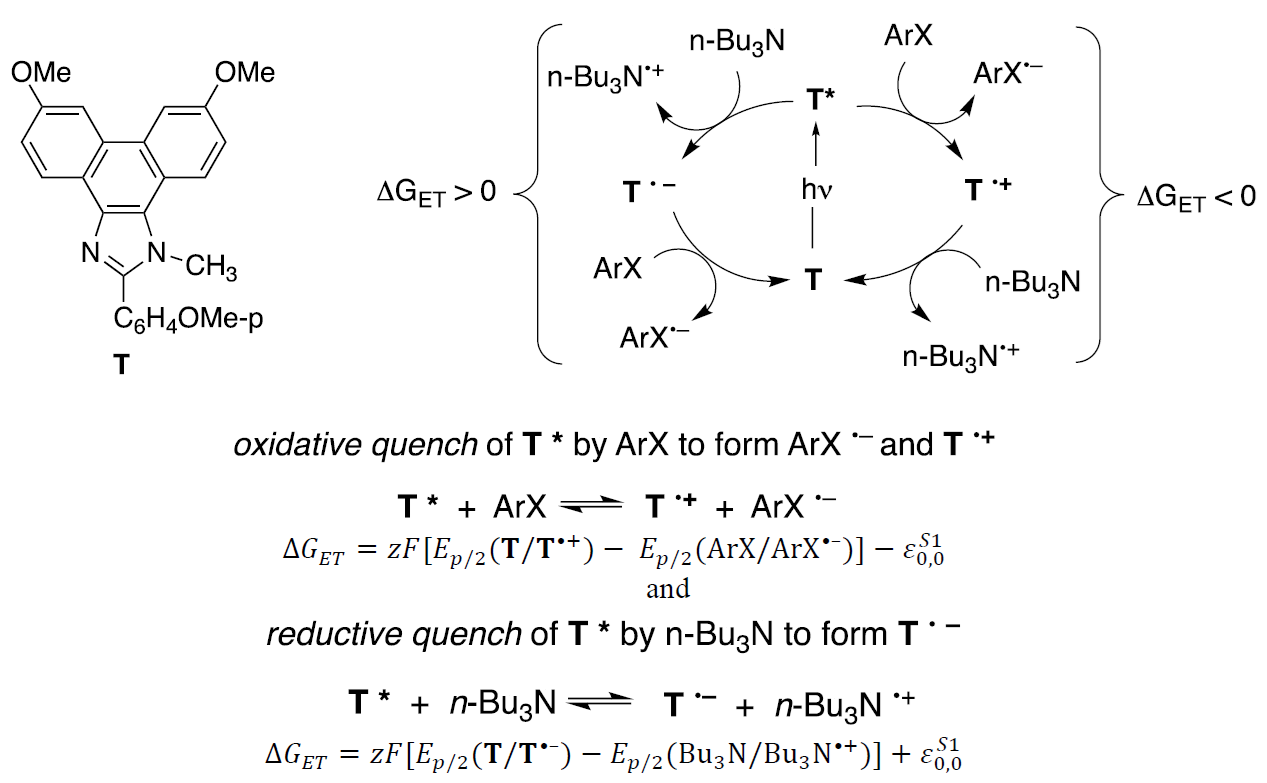
Fig. 4 Estimation of the Gibbs free energy for electron transfer. On the right, an oxidative quench leading to a Francke mediator cation radical. On the left, a reductive quench leading to a radical anion, T •-.
With this information in hand, we sought to exploit the excited state properties to achieve reduction, rather than oxidation that occurs electrochemically. The reactions were carried out using three 405 nm LEDs, n-Bu3N as a sacrificial electron donor and along with formic acid, a proton source. As illustrated by the transformations shown below, excellent results were obtained for reductive dehalogenation, borylation, and intramolecular cyclization. Eureka! The idea works!
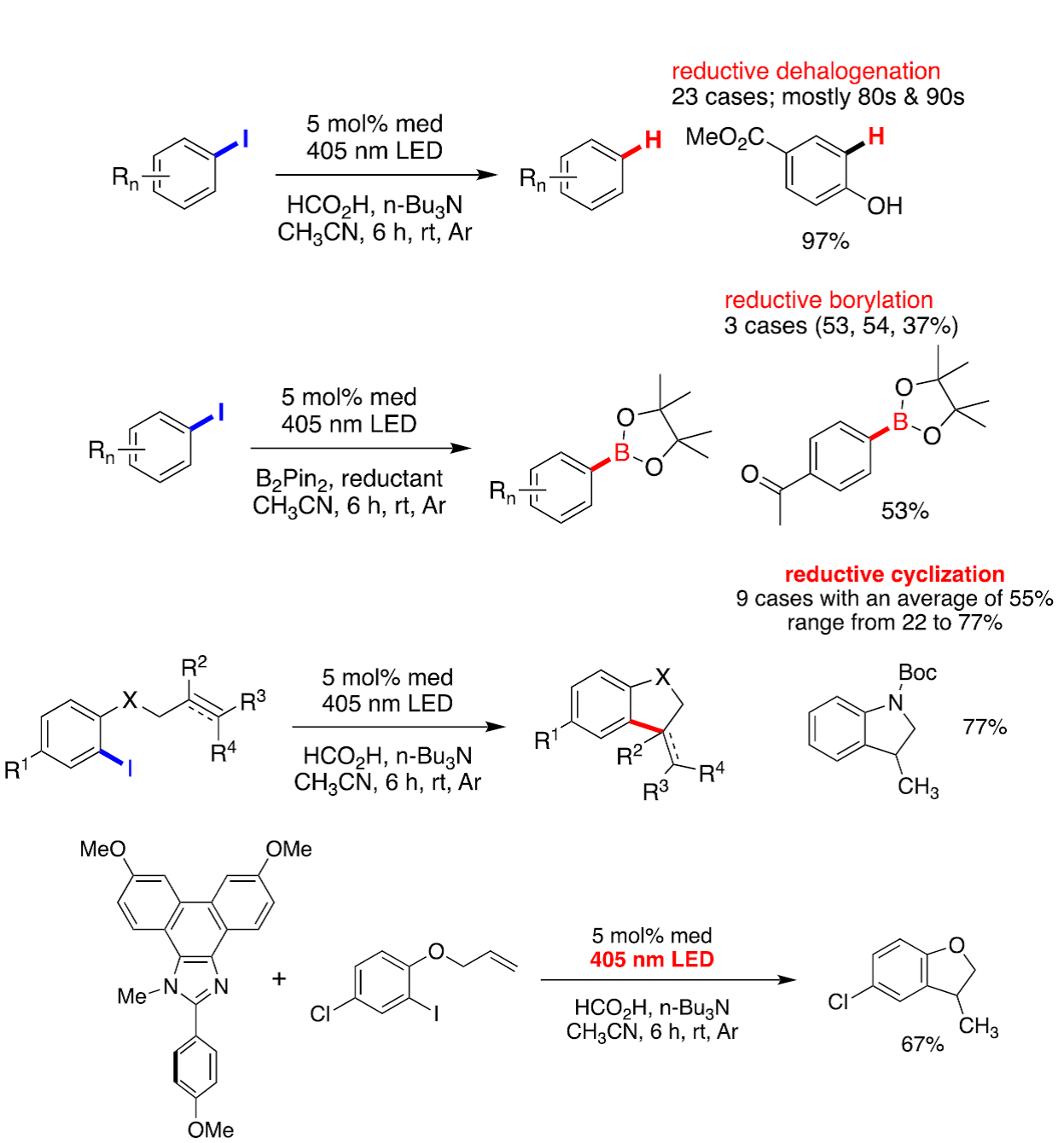
Examples of reductive dehalogenation (1st reaction), borylation (2nd reaction), intramolecular reductive cyclization (3rd and 4th reactions).
Clearly a single molecule, in this case a Francke mediator, can serve as an oxidizing agent or a reducing agent, with tunable properties, simply by changing the mode of activation! Are these concepts generalizable? Without a doubt, they are. There is much to be explored in what appears to be an exceptionally fertile territory, and researchers are challenged to do so.
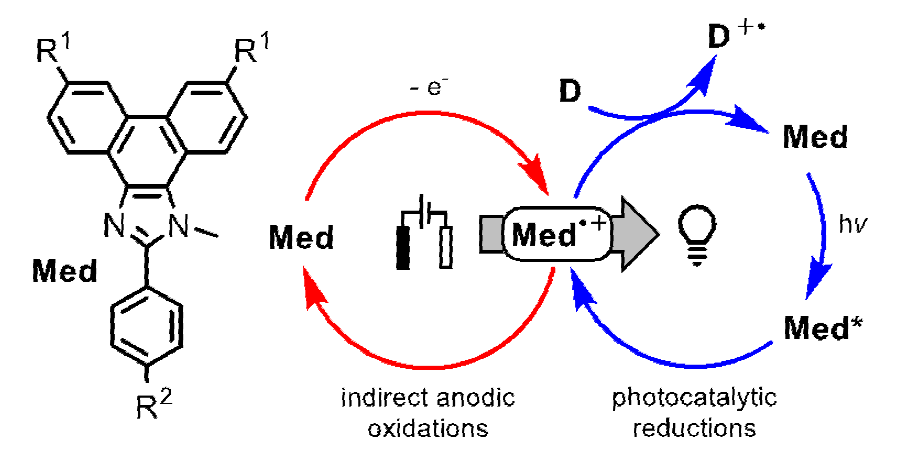
Let’s close by quoting from our recent manuscript, [14] ChemCatChem 2022, e202200830, https://doi.org/10.1002/cctc.202200830
“It should be highlighted that for the studied phenanthroimidazole system, only excitation by light irradiation enables the catalyst to reductively activate the targeted substrates, while electrocatalytic reductive transformations are not possible.” Electrochemically, oxidation occurs.
“In this context, the realization of the concept in the reverse direction, i.e., the transformation of an electrochemical mediator for reduction into a photooxidant, appears to be a worthwhile goal of future studies.” Hopefully, researchers will accept this challenge and explore what appears to be exceptionally fertile territory.

