











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkLas quemaduras son un escenario especial y complejo que se conceptualiza con base en los conocimientos actuales como disfunción cutánea aguda. Su fisiopatología es compleja e involucra la disfunción de múltiples órganos, que refleja una intrincada alteración molecular, subcelular, celular y de diferentes vías inmunometabólicas asociadas a una microcirculación y función endotelial alteradas. La hemostasia es parte de este involucro, lo que en el contexto de la quemadura favorece y perpetúa las disfunciones cutánea y orgánica en conjunto. Por lo anterior, es prioritario dentro del abordaje y manejo integral del enfermo quemado realizar una evaluación integral de la coagulación. El objetivo de esta revisión es poner al tanto al clínico de los conceptos actuales relacionados con las alteraciones de la hemostasia en el paciente quemado, su abordaje y alternativas terapéuticas.
Antecedentes
A partir del siglo XVIII y hasta la actualidad, el conocimiento acerca de la hemostasia se ha incrementado notablemente debido a que el avance de la ciencia y tecnología ha permitido una mejor comprensión de los procesos involucrados (Cuadro I).1-3
Cuadro I: Historia y evolución de la hemostasia y coagulación.
| Inicio | Filósofos y médicos griegos | Teoría del enfriamiento y del contacto con el aire. |
|---|---|---|
| 129-130 D.C. | Galeno | La sangre se aleja del corazón y se enfría |
| 1666 | Marcelo Malpighi | En su obra De Polypo Cordis: «Hay en la sangre una materia más abundante para formar la costra... tras repetidos lavados… todo el coágulo se vuelve pálido…» |
| 1731 | Jean Louis Petit | Describió los coágulos en las arterias lesionadas de individuos vivos; concluyó que participan en la detención de la hemorragia y que no sólo son la consecuencia del enfriamiento corporal que sigue a la muerte |
| 1832 | Johannes Müller | Afirma que la sangre no está animada debido a que sus células carecen de movimiento propio |
| 1836 | Alexander Buchanan | Comunica que el líquido mucinoso de los hidroceles no coagula de forma espontánea, sino que lo hace sólo al agregarle tejidos y suero. Así, aparece el concepto de que la coagulación podría ser un fenómeno en el que participan diversas sustancias |
| 1861 | Alexander Schmidt | Coagulación, requiere dos procesos: una sustancia proplástica o antecesora de la fibrina y una sustancia fibrinoplástica que promueve la conversión |
| 1903-1906 | Paul Morawitz | Propone que la coagulación de la sangre ocurre en dos etapas. La primera era la conversión de protrombina a trombina mediante la acción del factor tisular en presencia de calcio y la segunda, mediante la conversión de fibrinógeno a fibrina, gracias a la acción de la trombina |
| 1930 | Henrik Dam | Vitamina de la coagulación |
| 1930 | Armando Quick | Desarrolla la determinación del tiempo de protrombina o test de Quick |
| 1948 | Armando Quick y/Paul Owren | Describen la existencia del factor V (acelerina) y sexto o factor estable |
| 1949 | Andre De Vries | Factor VII, al que llamó proconvertina y convertina a su producto |
| 1954 |
Comité Internacional para la Nomenclatura de los Factores de Coagulación, por iniciativa de Irving Wright | Se aprobó la nomenclatura de los factores I a IX Se agregaron los factores X a XIII |
| 1958 | Robert MacFarlane y Oscar Ratnoff | Teoría de la cascada de la coagulación en la que habría dos vías, la extrínseca y la intrínseca, además de una vía común |
| 1964 | Shafer y Monroe | El complejo formado por el factor tisular y el factor VII no sólo participa en la activación del factor X, sino que también en la del factor IX |
| 1994 | Hoffman y cols. | Teoría celular de la coagulación, plantea que las superficies celulares controlan y dirigen el proceso de la hemostasia |
En 1964 Macfarlane y Ratnoff desarrollaron la teoría de la cascada de la coagulación en la que propusieron la presencia de dos vías: la extrínseca, constituida por el factor tisular y el factor VII, evaluada por el TP (tiempo de protrombina); y la intrínseca, en la que participan los factores XII, XI, VIII y V, que son proteínas con función enzimática que circulan en el plasma en su forma inactiva, como cimógenos, y que se activan por medio de la fragmentación de residuos de serina. Estas moléculas ya activadas son enzimas tipo serin proteasas, que fragmentarán residuos de serina de otra proenzima, activando de esta manera otro factor de la coagulación en una cadena progresiva. Ambas vías convergerán para activar al factor X, proceso que resulta en la transformación de protrombina a trombina y, a través de la trombina, el fibrinógeno en fibrina.4
Sin embargo, con este modelo, no se explican del todo las alteraciones de la hemostasia in vivo. Por lo tanto, se replanteó el modelo clásico de la cascada de la coagulación, con el objetivo de dar explicación a la mayoría de interrogantes que no pueden ser respondidas por este modelo. Hoffman y sus colaboradores propusieron en 2001 el modelo celular de la hemostasia, caracterizada por tres fases: iniciación, amplificación y propagación.5,6
El nuevo modelo, llamado teoría celular de la coagulación, plantea que las superficies celulares controlan y dirigen el proceso de la hemostasia; además, contempla el papel crucial de las plaquetas y otros elementos celulares que, de forma organizada y simultánea, generan trombina. A partir del nuevo modelo se complementa el entendimiento de los problemas clínicos observados en los diferentes trastornos de la coagulación.
Fisiología de la hemostasia
La hemostasia es un sistema fisiológico de defensa, tiene como principal función mantener la integridad vascular y evitar la pérdida hemática, además de tener una estrecha interrelación con el endotelio y la inflamación. Se activa por múltiples mecanismos que tienen en común la formación del trombo hemostático, reparación del daño y la posterior disolución del coágulo. El sistema de coagulación asegura la eficacia hemostática, mientras que el sistema fibrinolítico actúa como regulador. La hemostasia depende del equilibrio entre ambos sistemas.
El sistema de hemostasia se divide en dos mecanismos de respuesta:
Hemostasia primaria, caracterizada por la interacción entre el endotelio y las plaquetas; estas últimas participan a través de los procesos de adhesión, reclutamiento, activación y agregación para formar el tapón hemostático inicial.
Hemostasia secundaria o coagulación, donde participan los factores de coagulación que interaccionan sobre una superficie catalítica para formar una red de fibrina y, posteriormente, el coágulo.7,8
Hemostasia en quemaduras
Una quemadura mayor se define con base en el porcentaje de superficie corporal quemada, independientemente del grado de profundidad, con un involucro igual o mayor al 30% de superficie corporal total (SCT). La quemadura se asocia a un proceso inflamatorio que conduce a la activación del sistema de coagulación, consumo de factores de coagulación endógenos, activación de las vías procoagulantes, actividad fibrinolítica y el deterioro de la actividad de los anticoagulantes naturales.
Las alteraciones de la coagulación relacionadas a las quemaduras podrían dividirse en dos fases:
Coagulopatía primaria, en la fase temprana de la quemadura, debida a la activación de vías de procoagulación, con una mayor actividad fibrinolítica y deterioro de la actividad de los anticoagulantes naturales, lo que se asocia inherentemente al proceso inflamatorio. La relación entre la coagulación y la inflamación tiene una importante participación en la patogénesis de la lesión microvascular, con la consiguiente disfunción multiorgánica. Las anomalías del sistema de coagulación son más acentuadas durante la primera semana tras la lesión.
Coagulopatía secundaria, resultado de las intervenciones realizadas al paciente, tales como la reanimación hídrica, pérdidas hemáticas debido a la escisión de heridas y procesos infecciosos secundarios. La activación excesiva de la coagulación, junto con la activación de la inflamación resulta en trombosis microvascular y disfunción multiorgánica; este tipo de lesiones se asocia a la producción de una amplia gama de mediadores inflamatorios, como citocinas, que pueden inducir efectos inmunológicos relacionados a la gravedad de la lesión. Entre más extensa la quemadura, se acentúa la elevación temprana de interleucina 6 (IL-6) e interleucina 8 (IL-8), lo que se asocia a incremento en la mortalidad. En pacientes con quemaduras esta respuesta inmunoinflamatoria se relaciona con mayor índice de infecciones, disfunción orgánica y muerte (Figura 1).9-12
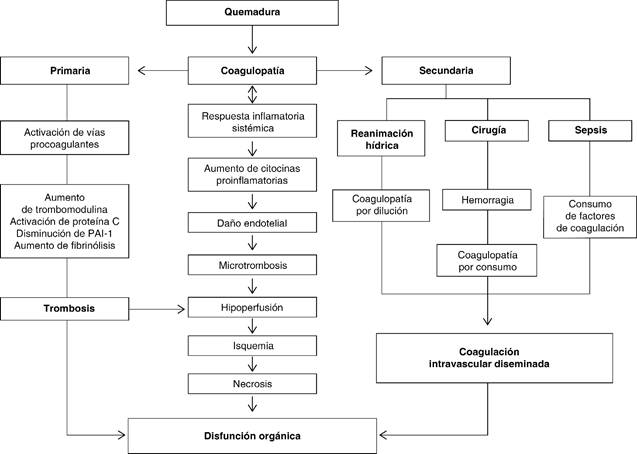
Figura 1: Flujograma en donde se observa la compleja interacción entre coagulopatía y disfunción multiorgánica en el enfermo quemado.
La coagulopatía asociada a quemaduras es secundaria a diferentes factores que se inician durante la generación de la quemadura, y se acentúan durante el proceso de reanimación y evolución del enfermo quemado.
Consumo. Los factores de coagulación y las plaquetas se consumen en la piel quemada o a nivel endotelial por lesión estructural.
Dilución. El desarrollo de coagulopatía temprana en pacientes con quemaduras graves puede ser consecuencia de hemodilución secundaria a una reanimación hídrica agresiva durante la fase de resucitación. La hemodilución generada, ya sea en estado de choque inicial o por pérdida sanguínea durante el tratamiento quirúrgico, produce un importante efecto en la hemostasia primaria. Un hematocrito bajo reduce el flujo axial, lo que condiciona que los eritrocitos fluyan en el centro de la arteriola y las plaquetas junto con el plasma sean desplazados en estrecha proximidad al endotelio; por lo tanto, se reduce la marginación de las plaquetas y la interacción célula endotelial-plaqueta. Posteriormente a la lesión tisular, existe un aumento en los niveles de hormonas, en especial, adrenalina y vasopresina, lo que asociado a la tormenta de citocinas condiciona diversos efectos sobre la hemostasia. La vasopresina estimula los cuerpos de Weibel-Palade, evento que tiene diversos efectos sobre la coagulación, de los que destacan la liberación del activador del plasminógeno tisular (t-PA), liberación del factor de Von Willebrand y la expresión de P-selectina en la superficie de las células endoteliales.
Mediadores inflamatorios. El factor de necrosis tumoral (TNF) e IL-1 inducen un cambio lento en el fenotipo de células endoteliales de un estado antitrombótico a un estado protrombótico, además de regulación a la baja de la trombomodulina y fibrinólisis (aumento del inhibidor del activador de plasminógeno-1 y expresión de glicosaminoglicanos a partir de la superficie de la célula, que limita la activación de la antitrombina. La lesión endotelial y la respuesta de fase aguda condicionan liberación de citocinas proinflamatorias como interleucinas 1, 6 y 8, interferón gamma y TNF alfa, además de hipovolemia, alteraciones en la contractilidad miocárdica e hipoperfusión tisular, que amplifican la coagulopatía de manera independiente y, en conjunto, perpetúan el daño. La hipoperfusión tisular favorece la expresión de trombomodulina y la activación de la proteína C, el efecto inhibidor de la proteína C activada en los factores de coagulación V y VIII, que junto con el inhibidor activador del plasminógeno 1 (IAP1), parecen ser claves para el desarrollo de coagulopatía secundaria a quemadura.
La deficiencia de antitrombina es resultado de la disfunción hepática, incremento del consumo durante la coagulación intravascular diseminada (CID), síndrome de respuesta inflamatoria sistémica (SIRS) y sepsis. En un estudio de 201 pacientes quemados, se evaluó la incidencia de deficiencia de antitrombina III; 108 de ellos desarrollaron deficiencia durante su hospitalización; el porcentaje de pacientes que presentaron deficiencia fue mayor en los primeros cinco días después de la lesión, siendo mayor el riesgo de presentar dicha deficiencia en pacientes con mayor porcentaje de quemadura y presencia de lesión por inhalación, lo que se asoció a incremento en la estancia hospitalaria y mortalidad.13-16
La coagulopatía del enfermo quemado tiene una presentación clínica heterogénea, que va de un estado hipercoagulable a la CID. De acuerdo al estudio consultado, la incidencia de cada una de éstas es variable, lo que puede estar en relación con la diferencia en los criterios diagnósticos empleados. Lo importante en este aspecto es tenerlas en cuenta para hacer un diagnóstico temprano y oportuno e implementar la mejor estrategia terapéutica.17-19
Definiciones
Se han adoptado muchos términos para definir la coagulopatía en los pacientes con quemaduras severas, pero la terminología más empleada es:20,21
Estado de hipercoagulabilidad sistémica en el quemado, que se caracteriza por niveles elevados de factor activado VII, complejo trombina-antitrombina (CTAT), productos de degradación del fibrinógeno y del inhibidor del activador del plasminógeno tipo 1(IAP-I), en combinación con disminución de los niveles de antitrombina y otros anticoagulantes endógenos.
Coagulopatía intravascular diseminada en quemaduras, que se diagnostica con base en los criterios de la Sociedad Internacional de Coagulación y Trombosis (ISTH). Su fisiopatología es semejante a la de los pacientes con sepsis o trauma grave, caracterizada por la activación intravascular de la coagulación, de las vías del sistema antifibrinolítico, el consumo de los factores de coagulación y proteínas reguladoras de la coagulación, que condiciona depósitos de fibrina en la microcirculación, con la consecuente hipoperfusión microcirculatoria y disfunción multiorgánica. La fase de consumo se caracteriza por una acentuada activación de la fibrinólisis, consumo de fibrinógeno y hemorragia.
Coagulopatía aguda en el quemado. Se propuso con base en la medición de la relación de normatización internacional (INR) o en el tiempo de tromboplastina parcial activada (TTPa) medido al ingreso del paciente al hospital, y se basa en la presencia de un INR o TTPa anormal dentro de las primeras 24 horas tras la quemadura, con valores de INR de ≥ 1.3 o un TTPa de ≥ 1.5 veces lo normal.22
Diagnóstico
La evaluación diagnóstica de la coagulopatía en pacientes quemados es compleja y se fundamenta en la evaluación de la coagulación con base en diferentes exámenes realizados en el laboratorio de coagulación, de los que destacan:
a) Pruebas globales de coagulación
Las pruebas de coagulación que se utilizan con mayor frecuencia son el tiempo de protrombina (TP), tiempo de tromboplastina parcial activado (TTPa), tiempo de trombina (TT) y el nivel sérico de fibrinógeno, que se consideran dentro de las pruebas globales. Las pruebas más específicas deben incluir medición de dímero D y determinación del factor de Von Willebrand.
El TP determina la función del factor VII y los factores X y V, protrombina y fibrinógeno. Además de evaluar los estados de deficiencia congénita de factores VII, X, V y protrombina. El TP también se utiliza para medir la anticoagulación con antagonistas de la vitamina K (warfarina y acenocumarina).
El TTPa evalúa la función de los factores XII, XI, IX y VIII, precalicreína y cininógeno de alto peso molecular. El TTPa es útil para identificar hemofilia A (deficiencia del factor VIII), hemofilia B (deficiencia del factor IX, de la enfermedad de Christmas), deficiencia del factor XI (hemofilia C o la enfermedad de Rosenthal), así como la medición de la heparina no fraccionada y los inhibidores directos de la trombina argatrobán, bivalirudina y lepirudina.23,24
El TT evalúa el estado funcional del fibrinógeno, en el que el TP y el TTPa son menos sensibles. Es muy sensible a la heparina y a los inhibidores directos de trombina.25,26
Cuando los resultados de las pruebas de coagulación de un paciente son anormales, es importante determinar si el resultado anormal se debe a un estado de deficiencia, a la presencia de un inhibidor o a un estado fibrinolítico acentuado, lo que determinará la mejor estrategia a seguir, para afinar aún más el diagnóstico o planear el tratamiento.
El dímero D es un fragmento de fibrina reticulada que se produce por la lisis de la fibrina. Es un marcador de trombosis y fibrinólisis, útil para el diagnóstico de CID o trombosis aguda.
b) Cuenta y funcionalidad plaquetaria
La determinación cuantitativa de las plaquetas se realiza rutinariamente en la biometría hemática, en donde se analiza con precisión el número y tamaño plaquetario en un volumen determinado. El tiempo de sangrado es una prueba sencilla que se realiza a la cabecera del enfermo; nos indica de manera global la funcionalidad plaquetaria.
Existen otras pruebas específicas como la determinación de la fracción inmadura de plaquetas (FIP), una medida del porcentaje de plaquetas jóvenes en la sangre, el tiempo de sangrado (TS), el análisis de la función plaquetaria y los estudios de agregación de plaquetas. El volumen medio de plaquetas es útil porque un gran volumen medio de plaquetas sugiere la presencia de un gran número de plaquetas inmaduras (las plaquetas jóvenes son más grandes).
El porcentaje de la FIP se evalúa por tinción de las plaquetas con un colorante fluorescente, el cual se puede medir con analizadores automatizados debidamente equipados con citometría de flujo. Los pacientes con mayores porcentajes de plaquetas jóvenes presentan incremento en la producción de plaquetas, lo que sugiere la presencia de un trastorno que causa consumo de plaquetas/destrucción.
El análisis de la función plaquetaria (AFP-100) evalúa mediante la aspiración de la sangre entera a altas velocidades de cizallamiento a través de las membranas perforadas recubiertas con colágeno y epinefrina o colágeno y difosfato de adenosina (ADP). El AFP-100 será anormal en pacientes con trombocitopenia (recuento de plaquetas < 80-100.000/l), por lo que el resultado de esta prueba no se puede utilizar para evaluar la función plaquetaria en pacientes con recuentos de plaquetas en o por debajo de este nivel. Sin embargo, en un paciente con hemorragia aguda y recuento plaquetario normal, el AFP-100 indica una deficiencia adquirida (por ejemplo, aspirina, uremia, etcétera) o un trastorno congénito de las plaquetas (por ejemplo, el síndrome de Bernard-Soulier). Las pruebas de agregación de plaquetas evalúan la función de las plaquetas mediante la exposición de plasma rico en plaquetas o sangre entera a diferentes agonistas plaquetarios tales como trombina, epinefrina, ADP, y ristocetina. La trombocitopenia (plaquetas < 50,000/l) dará lugar a resultados anormales.27-30
c) Sistema fibrinolítico
El sistema fibrinolítico se puede evaluar mediante la determinación de la concentración y la función de las proteínas fibrinolíticas, tales como plasminógeno, activador tisular del plasminógeno, alfa2-antiplasmina y activador del plasminógeno 1. La reducción en la alfa2-antiplasmina y PAI-1, dos proteínas que contrarregulan a la plasmina (enzima fibrinolítica) y al activador tisular del plasminógeno, acentúa el estado fibrinolítico.16,31-34
d) Pruebas viscoelásticas
La tromboelastografía (TEG) y tromboelastometría rotacional (ROTEM) son pruebas de viscoelasticidad que evalúan la formación y disolución del coágulo. Analizan la función global de las proteínas de la coagulación, las plaquetas y el sistema fibrinolítico. TEG y ROTEM han sido utilizadas como pruebas para identificar pacientes con alteración en la coagulación, la función plaquetaria o fibrinólisis. Han tenido relevancia en la evaluación de la coagulación en el trasplante hepático y cirugía cardiaca; sin embargo, investigaciones actuales justifican su aplicación más amplia, como en enfermos con trauma, sepsis y hemorragia obstétrica. En el paciente quemado, la ROTEM en especial, tiene una excelente área de estudio, sobre todo para la evaluación global de la coagulación durante la reanimación o el desarrollo de complicaciones, lo que la posiciona como una importante herramienta para guiar no sólo el monitoreo de la coagulación, sino también el tratamiento a implementar.35-38
Tríada letal
La tríada de hipoxia, acidosis e hipotermia acentúa la coagulopatía e incrementa el riesgo de hemorragia y mortalidad. La hipotermia y la acidosis, generadas principalmente por un estado de choque acentuado y la pérdida de temperatura a nivel cutáneo, afectan negativamente la capacidad funcional de las plaquetas y las proteasas de la coagulación.39,40
La temperatura central menor a 35 oC se ha asociado a disfunción plaquetaria, desregulación de factores de coagulación y del sistema de fibrinólisis. Una temperatura corporal de 34 oC puede afectar la actividad de la coagulación en 80%, además de asociarse a mayor índice de transfusiones y, por lo tanto, una mayor morbilidad y mortalidad que asciende hasta 80%.
Las alteraciones hemostáticas son más evidentes en pH menor de 7.1 y temperaturas por debajo de 33 oC. En modelos animales, la acidosis exacerba la hemorragia, no sólo por una disminución en la generación de trombina, sino también mediante la aceleración de la fibrinólisis.
Lavrentieva identificó el desarrollo de coagulopatía como un predictor independiente de mortalidad a 28 días en pacientes con quemaduras graves.41,42 Los niveles bajos de antitrombina también son predictor independiente de la mortalidad y la estancia intrahospitalaria. La coagulopatía en los pacientes quemados se asocia con un mayor requerimiento de transfusiones, estancia en la UTI, días de ventilación mecánica y riesgo de disfunción multiorgánica. Por otro lado, existe una evolución dinámica de un estado de coagulopatía y hemorragia a uno procoagulable, lo que se manifiesta con un riesgo elevado de desarrollar enfermedad tromboembólica venosa.43-45
Tratamiento
No hay directrices claras o recomendaciones bien definidas sobre el tratamiento de la coagulopatía en pacientes con quemaduras graves, pero tomando en cuenta que las quemaduras se incluyen dentro del trauma y que la fisiopatología de la coagulopatía es similar, se pueden extrapolar algunas de las que se siguen para el manejo del trauma en general.
De estas recomendaciones destacan:
Reanimación temprana con base en objetivos y evitando la sobrerreanimación, lo que permitirá una mejor perfusión tisular, limitando la lesión endotelial y la dilución y consumo de factores de coagulación.
Monitorización de la coagulación desde el ingreso del enfermo al hospital o la UTI.
Oxigenación tisular, reanimación con cristaloides y manejo de temperatura, lo cual incluye oxigenación tisular y mantener una presión arterial media de 80-90 mmHg; en enfermos que cursen con lesión del sistema nervioso central se recomiendan presiones de perfusión > 80 mmHg. Se aconseja limitar el uso de solución salina 0.9%, restringir coloides. La albúmina se podrá emplear posteriormente a las cinco horas de la lesión, en especial en aquellos enfermos que requieran de dosis elevadas de cristaloides para mantener un adecuado estado hemodinámico. Es importante tener en mente que se puede inducir coagulopatía en 40% de los enfermos que reciben dos litros de cristaloides en infusión y en más de 70% de aquéllos que reciben más de cuatro litros. Los coloides basados en almidón acentúan más la coagulopatía por su interacción con el fibrinógeno y el factor de Von Willebrand, alterando la polimerización de la fibrina y la estabilidad del coágulo. En diferentes estudios se ha mostrado que la vasodilatación posterior a trauma y/o quemaduras, por la respuesta inflamatoria que inducen, acentúa la vasodilatación, lo que incrementa de manera significativa los requerimientos de líquidos; para limitar este efecto se aconseja el uso temprano de vasopresores. Es recomendable mantener la temperatura corporal entre 36 y 37 grados centígrados durante la reanimación, para lo que es necesario utilizar soluciones tibias y elevar la temperatura de la habitación.
Control de la hemorragia, ya sea con cirugía de control de daños, medidas hemostáticas locales y/o radiología intervencionista, acciones que limitarán el requerimiento de líquidos y el uso excesivo de productos sanguíneos.
Monitoreo y manejo de la coagulopatía. El monitoreo de la coagulación a partir del ingreso del enfermo es fundamental, lo que permitirá un diagnóstico preciso de la coagulopatía y, en especial, dirigirá de manera objetiva su tratamiento y el uso de productos sanguíneos y fármacos. En caso de hemorragia crítica aguda, se recomienda el uso de concentrados eritrocitarios y plasma fresco en relación 1:2, asociado al empleo de antifibrinolíticos, ya sea ácido tranexámico o ácido épsilon aminocaproico. Es importante recordar que la anemia predispone a mayor hemorragia, por lo que con base en las recomendaciones actuales, se deberá considerar mantener niveles de hemoglobina entre ocho y 10 g/dL.
El concentrado de fibrinógeno y los crioprecipitados están indicados cuando la hemorragia es significativa y hay evidencia cuantitativa, o por pruebas viscoelásticas de disminución en los niveles de fibrinógeno. Es importante tomar en cuenta que las recomendaciones actuales proponen mantener niveles de fibrinógeno entre 1.5 y 2 g/dL. Este objetivo se puede alcanzar con el empleo de concentrado de fibrinógeno a dosis inicial de tres, que equivale de 15 a 20 unidades de crioprecipitados. Nuevas dosis se decidirán con base en la respuesta clínica y el seguimiento de niveles de fibrinógeno o pruebas viscoelásticas. Es conveniente no olvidar la cuenta y funcionalidad plaquetaria; se recomienda mantener cifras por arriba de 50,000/mcL; en caso de un mayor descenso y/o disfunción, está indicado el empleo de aféresis plaquetaria o concentrados plaquetarios.
El concentrado de complejo de protrombina (CCP) está constituido por factores de coagulación en dosis elevadas que se conceptualizan como fármacos indicados para el manejo de diferentes coagulopatías, en especial cuando existe intoxicación con antagonistas de vitamina K; últimamente se ha considerado en conjunto con el concentrado de fibrinógeno para el manejo de coagulopatía en trauma.46

