










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 no.2 Ciudad de México abr./jun. 2004
Investigación
Quantum Mechanical Approach to Leucine + OH Gas Phase Reaction. Mechanism and Kinetics
Annia Galano,*1 J. Raúl Álvarez-Idaboy,1 Esther Agacino2 and Ma. Esther Ruiz-Santoyo1
1 Instituto Mexicano del Petróleo, Eje Central Lázaro Cárdenas 152, 007730, México D. F., México. Phone: (+52 55) 9175 6949, Fax: (+52 55) 9175 6935. E-mail: agalano@imp.mx; jidaboy@imp.mx
2 Centro de Investigaciones Teóricas, Facultad de Estudios Superiores Cuautitlán, Universidad Nacional Autónoma de México, Cuautitlán Izcalli, CP 54740, Edo. Méx., México.
Recibido el 18 de febrero del 2004.
Aceptado el 10 junio del 2004.
Abstract
The gas phase OH hydrogen abstraction reaction from leucine has been studied using Density Functional Theory (B3LYP) calculations and the 6-311G(d,p) basis set. The structures of the different stationary points are discussed. Reaction profiles are modeled including the formation of pre-reactive complexes, and negative net activation energy is obtained for the overall reaction. BSSE corrections are included. A complex mechanism is proposed, and the rate coefficients are calculated using Transition State Theory over the temperature range 250 - 350 K. The rate coefficients are proposed for the first time and it was found that in the gas phase the hydrogen abstraction occurs almost exclusively from the gamma site. The following expressions, in L⋅mol-1⋅s-1, are obtained for the alpha and gamma H-abstraction channels, and for the overall temperature dependent rate constants, respectively: kα = (2.19 ± 0.05) x 107 exp[(905 ± 12)/T], kγ = (3.19 ± 0.05) x 107 exp[(2450 ± 10)/T], and ktot = (3.80 ± 0.08) x 107 exp[(2378 ± 12)/T].
Key words: leucine, OH radical, H abstraction, rate, mechanism.
Resumen
Se estudia la reacción de abstracción de hidrógeno de leucina por OH en fase gaseosa usando cálculos de la teoría de funcionales de la densidad (BYLYP) y el conjunto de bases 6-311G(d,p). Se discuten las estructuras de los diferentes puntos estacionarios. Se modelaron los perfiles de reacción, incluyendo la formación de complejos pre-reactivos, y se obtuvo una energía de activación negativa para la reacción completa. Se incluyeron correcciones BSSE. Se propone un mecanismo de reacción complejo, y se calcularon los coeficientes de velocidad usando la teoría del estado de transición en el rango 250-350 K. Se proponen por primera ocasión los coeficientes de velocidad y se encontró que en fase gaseosa, la abstracción del hidrógeno procede casi exclusivamente de la posición gama. Las siguientes expresiones, en L mol-1 s-1, se obtienen de los canales de abstracción alfa y gama, respectivamente, y para las constantes de velocidad dependientes de la temperatura: kα = (2.19 ± 0.05) × 107 exp [ (905 ± 12)/T], kγ = (3.19 ± 0.05) × 107 exp [ (2450 ± 10)/T], and ktot = (3.80 ± 0.08) × 107 exp [ (2378 ± 12)/T].
Palabras clave: leucina, radical OH, abstracción de H, velocidad, mecanismo.
Introduction
Leucine is an essential amino acid that is found as a structural element in the interior of proteins and enzymes. It is a member of the branched-chain amino acid (BCAA) family. Leucine is the third most abundant amino acid and its frequency of occurrence in proteins is 7.5% [1]. Leucine has two methyl groups in the side chain attached to Cγ.
Oxidative damage to proteins has been involved in several pathological disorders and it is generally initiated by OH radicals, which may be formed intracellularly by a Fenton type reaction, by Haber-Weiss recombination, via water radiolysis, or by other radicals created from enzyme reactions [2-6]. OH radicals can also be produced by ultraviolet and ionizing radiations [7]. In addition, most amino acids and their derivative synthesis have been based on ionic procedures in the past, but recently, the discovery that radical reactions can be performed with a high degree of regio and stereo control [8-10], has contributed to the increased interest in this kind of reactions.
Goshe and coworkers [11-12] have studied the sites of OH radical reaction with some amino acids solution, by EPR spin trapping and 2H NMR detection of induced 1H/2H exchange. They reported that methine and methylene positions are more reactive than methyl positions, and that this reactivity is reduced by their closer proximity to the α-amino and carboxylate groups. The authors also suggest that knowledge of the reactivities of the amino acid C-H bonds with OH is required to predict sites of biological oxidative damage to proteins.
However, as far as we know, there is no study of the proportion in which the different possible leucine radicals may be formed. Logically, the chemical behavior of an amino acid in its free form and in a peptide chain is not the same, because it depends on the chemical environment. However, it is reasonable to assume that for those sites in the side chain, the closer to the alpha carbon, the more affected by the peptidic surroundings. A reactivity study should begin with an analysis of the intrinsic reactivity, due to its strong influence in the reactivity that the amino acid will exhibit in solution or in a peptidic environment. Following this way of thinking, the attack of an OH radical to a protein can be considered as an attack to one of the amino acids in it. This reaction is postulated as the first step of a complex mechanism, which ends with the fragmentation of the peptide chain of the protein. The mechanism of such a reaction is not completely understood.
To our knowledge, neither activation energies nor rate constants have been reported for the leucine + OH reaction in gas phase. There is also a lack of knowledge about the transition structures and the mechanism, as well as about the temperature dependence of the rate coefficients. Therefore, the objective of this work is to provide quantitative information about the mechanism and kinetics of this reaction, which can be considered as the very first step of the oxidative damage. Two different channels have been modeled corresponding to H abstractions from the alpha and gamma sites. The abstractions from the terminal CH3 and from beta CH2 were not included, based on previous finding [13-16] that the abstraction of those H atoms should be neglected compared to the other two possible channels.
The reaction paths were modeled taking into account the results of previous studies performed by our group on similar systems. According to references [13-16], it is a reasonable assumption that the amino acid + OH reactions occur via a two step mechanism, with a reversible first step leading to a pre-reactive complex, and a second step yielding the corresponding radical and H2O (for more details on the mechanism see reference 17).
Computational Details
Electronic structure calculations have been performed with the Gaussian 98 [18] program. Full geometry optimizations were made for all the stationary points using the B3LYP method and the 6-311G(d,p) basis set. Restricted calculations were used for closed shell systems and unrestricted ones for open shell systems. Frequency calculations were carried out for all the stationary points at the corresponding level of theory. Local minima and transition states were identified by the number of imaginary frequencies (NIMAG = 0 or 1, respectively). In addition, the vibrational mode with imaginary frequency was inspected using the GaussView program, and it was confirmed that it does connect reactants and products. Zero point energies (ZPE) were included in the determination of energy barriers and thermal corrections to the energy (TCE) were included in the heats of reaction calculations.
Basis Set Superposition Error (BSSE) has been included. The relevance of the BSSE has been well established from a theoretical point of view [19,20] and its utility has been proved in systems relatively similar to the amino acids [21]. However the counterpoise method [22,23] of calculating it has often been criticized [24,25]. B3LYP methods show a tendency to underestimate the activation energies for molecule + radical reactions and the inclusion of the BSSE correction would improve the correspondence with experimental results.
The Transition State Theory (TST) methodology [26,27], implemented in the Rate 1.1 program [28], was used to calculate the rate coefficients since it has the advantage of being non-expensive for high level ab-initio calculations.
Results and Discussion
Molecular Structures
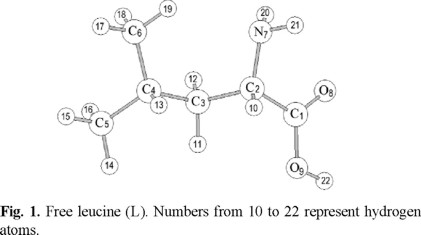
Leucine is an aliphatic amino acid, with a methyl group linked to the gamma carbon. It was fully optimized at B3LYP/6-311G(d,p) level of calculation and an intramolecular interaction was found between N7 and H19 (Fig. 1), with the interaction distance equal to 2.47 Å. In this figure the carbon atoms numbered as 2, 3 and 4 correspond to the sites known as alpha, beta and gamma, respectively. A Bader topological analysis [29] of the B3LYP/6-311G(d,p) wave function was performed in order to confirm the existence of the interaction. The electronic charge density (ρ) at the bond critical point was found to be ρ7-19 = 0.0134, and the Laplacian of ρ, ∇ 2ρ7-19 = -0.0100, confirming the existence of the interaction.
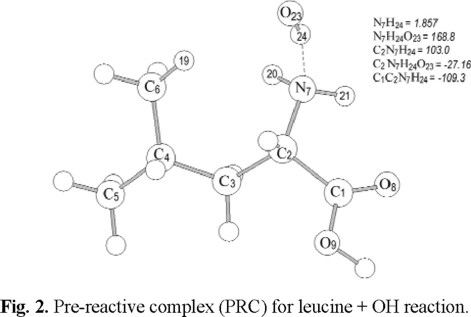
Since two reaction paths have been considered, the hydrogen abstraction from the alpha, and gamma carbons, two reactant complexes (RC) were modeled from the optimized transition structures. For each channel an Intrinsic Reaction Coordinate (IRC) calculation was performed and the final structure towards the reactants side was fully optimized as a minimum. The reactant complexes obtained this way for the alpha and gamma channels are identical and show the leucine structure almost identical to the free molecule (Fig. 2). The main differences between the RC and the free molecule are a slight elongation (0.01 Å) of the N7-C2 bond length and the dihedral angles associated to the NH2 group (C4C3C2N7 = -84.8o, C1C2N7H20=134.0o, C1C2N7H21=15.5o). The formation of the studied reactant complexes is caused by the interaction between the H atom in the OH radical and the N atom in leucine, the distance between them being 1.86 Å, which corresponds to a hydrogen bond interaction distance. This interaction provokes the torsion of the amino group, described above, and its bond critical point is characterized by ρ7-24 = 0.0386, and ∇ 2ρ7-24 = -0.0247, according to Bader's topological analysis. An additional interaction was also found between the O atom in the OH radical and one of the hydrogens in leucine, with O23-H19 distance of 2.91 Å, ρ19-23 = 0.0042, and ∇ 2ρ19-23 = -0.0035.
The fully optimized geometries of the transition states corresponding to alpha (TSα) and gamma (TSγ) H-abstractions are shown in Figure 3. Two major changes occur due to the TSα formation, the C2-H10 bond elongation by 0.05 Å and the C2-N7 bond shortening by 0.01 Å. The H-attack on the alpha site was found to be non-collinear, with the C2H10O23 angle, equal to 145.9º. An attractive interaction was found between the O atom in the OH radical and H19 atom in the amino acid, with the distance N19H23 equal to 2.43 Å. Due to this attractive interaction the optimized TSα shows a ring like structure (Fig. 3). The interaction was analyzed by the Bader's topological analysis, and a bond critical point was found, with ρ19-23 = 0.0110, and ∇ 2ρ19-23 = -0.0081.
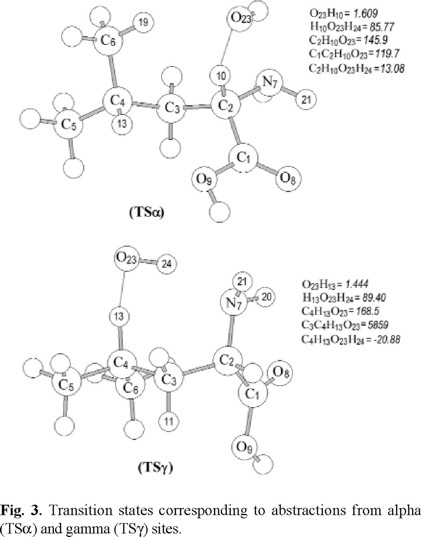
The main structural change associated to the TSγ formation (Fig. 3) is the C4-H13 bond elongation by 0.06 Å. The attack of the OH radical on the gamma site was found non-collinear, with the C4H13O23 angle equal to 168.5o. For this channel it was found an attractive interaction between the H atom in the OH radical and the N atom in leucine, with the N7H24 distance equal to 1.97 Å and showing a seven members ring like structure. The topological analysis leads to a bond critical point with ρ7-24 = 0.0307, and ∇ 2ρ7-24 = -0.0218. Comparing these values with those corresponding to TSα, it can be seen that in TSγ there are a stronger interaction and a larger stabilization than in TSα.
According to the fully optimized transition structures discussed above, the stabilizing ring- like arrangement seems to be feasible for other than alpha transition states in amino acids with aliphatic side chains. They would just differ in the atoms forming the ring, and their number logically depends on the abstraction site.
The distance between the O atom in the OH radical and the H to be abstracted was found to be equal to 1.61 Å in TSα and 1.44 Å in TSγ, indicating that the first one is an earlier transition state and suggesting a lower energy barrier and a process more likely to occur. This leads an expectation in the opposite direction than the stabilizing hydrogen bond like interaction discussed above. Thus, based on the hydrogen bond like stabilization in the transition structures, it should be expected the gamma H-abstraction channel energy barrier to be lower than that of the alpha channel. Conversely, it was found that TSα is an earlier transition state, which suggests a lower barrier and a process more likely to occur. Therefore, energetic and kinetics analyses are necessary to determine which of these channels is predominant in the H abstraction from leucine.
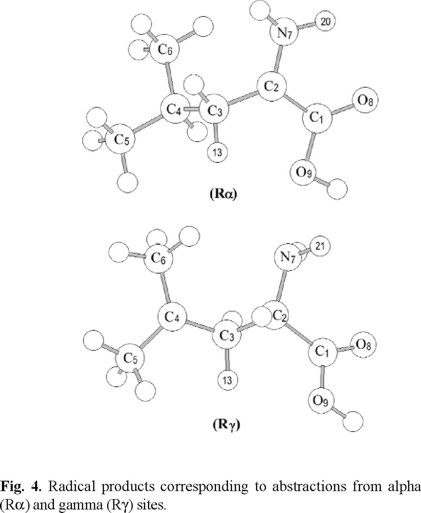
The structures of the radical products due to the alpha (Rα) and gamma (Rγ) hydrogen abstractions are shown in figure 4. Rα shows the expected planar section around the alpha carbon and the shortening of the C1-C2, C2-C3 and C2-N7 distances by 0.16, 0.05 and 0.02 Å, respectively, compared to the free leucine. Rγ presents a planar section around the gamma carbon (C4) and the shortening of the C3-C4, C4-C5 and C4-C6 distances by 0.06, 0.06 and 0.07 Å, respectively.
Energies and Mechanisms
The heats of reaction (∆Εreac) including thermal corrections to energy (TCE) at 298 K, and the L parameters were calculated for the two channels (Table 1). The L parameter denotes if a transition state structure is reactant-like (L < 1) or product-like (L > 1) and also quantifies the corresponding trend. Consequently, there must be a direct relation between the L value and the heat of reaction of a specific pathway, according to Hammond's postulate [30]. L parameters were calculated for each modeled channel, following references 31 and 32, as:
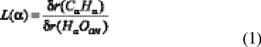
and

In these equations, δr(CαHα) and δr(CγHγ) represent the variation in the breaking bond distance between transition states and reactants, along the different channels, while δr(HαOOH) and δr(HγOOH) represent the variation in the forming bond distance between transition states and products.
The direct relation between L parameter values and the heats of reactions is very well accomplished in the leucine + OH reaction. In addition, there is a consistent pattern for the value of these two magnitudes and the nature of the abstraction site. The alpha abstraction shows a very low value of L, about 0.07, and a ∆Εreacc = -41.49 kcal/mol, being the most exothermical channel, while for the gamma channel L = 0.13 and ∆Ereacc = -20.12 kcal/mol.
The formation of the pre-reactive complex leads to a stabilization energy of 2.97 kcal/mol, suggesting that the corresponding mechanism is complex. The reaction path has been modeled by two steps, the first one leading to the formation of the pre-reactive complex (or reactant complex, RC), and the second one yielding the corresponding radical and water:
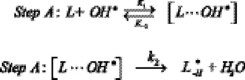
In figure 5, the energies of four critical points of the potential surface, relative to reactants, have been represented for each abstraction channel. They are: the isolated reactants, the pre-reactive complex, the transition states and the isolated products. The energy profiles show that the energies of all the studied stationary points are lower than the energy of the reactants. Therefore, a negative apparent energy barrier (Eapp = ETS -ΣEreac) was found for both channels, the gamma abstraction transition state being 0.91 kcal/mol lower than the isolated reactants and 1.53 kcal/mol lower than that of the alpha channel (Table 1). The existence of a reactant complex, about 3 kcal/mol more stable than the isolated reactants, justifies the gamma Eapp sign and leads to a complex mechanism, characterized by a reversible first step with no energy barrier and a non-reversible second step with barriers of 2.06 and 0.53 kcal/mol for the alpha and gamma channels, respectively (Fig. 5).
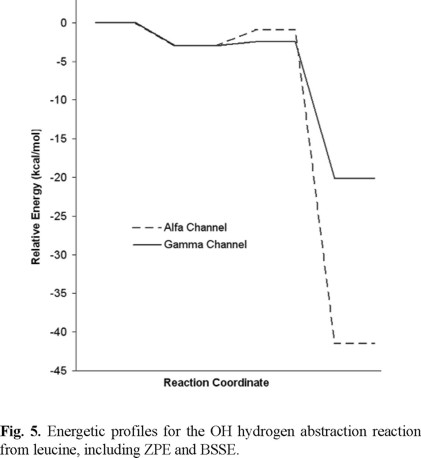
According to the reaction profiles features it can be assumed that because of its lower energy barrier the gamma H-abstraction channel is the most likely to occur, regardless of the larger exothermicity of the alpha channel. This assumption can be made based on the fact that heats of reaction mainly depend on the strength of the breaking and forming bonds, while the barriers also include dynamic effects in the transition structures, like the intramolecular stabilization discussed above.
Rate coefficients
We have assumed that the reaction occurs according to a two step mechanism involving a fast pre-equilibrium between reactants and the pre-reactive complex, followed by the elimination of a water molecule. If k1 and k-1 are the forward and backward rate constants for the first step, and k2 corresponds to the second step then, a steady state analysis leads to a rate coefficient for the overall reaction (k) of each channel. The mathematical expression for k can be written as:
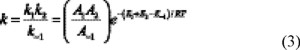
In equation 3 it has been assumed that k-1 is much larger than k2. According to the difference ETS - ER, this assumption can be true only if the pre-exponential factor (A) of k-1 is at least two orders of magnitude larger than that of k2. The rate constant of the step 1 reverse reaction, which goes from reactant complex to isolated reactants, can not be calculated within the TST frame, but it can be considered as a dissociation reaction. According to the data reported in the NIST Chemical Kinetics Database [33] the pre-exponential factor for this kind of reaction is about 1015 L mol-1 s-1, while the pre-exponential factor calculated in this work for step 2 is about 1011 L mol-1 s-1. These values make A-1 about four orders larger than A2 and validate the assumption made in equation 4. The larger value of A-1 can be explained by the activation entropy (∆S≠), which is small and negative for step 2 because the transition state structure is tighter than the reactant complex, while ∆S≠-1 is large and positive because six vibrational degrees of freedom are being converted into three translational plus thee rotational degrees of freedom. The assumption that k-1 is much larger than k2 was proposed for a similar system by Singleton and Cvetanovic [34].
Since E1 is equal to zero (see Figure 5), the apparent energy barrier (Eapp) for the overall reaction is:
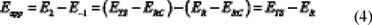
where ER, ERC and ETS are the total energies of the reactants, pre-reactive complex and transition state, respectively.
Applying basic statistical thermodynamic principles, the equilibrium constant of the first step can be expressed in terms of the partition functions (Q) as:

and k2 can be obtained from TST in the form:
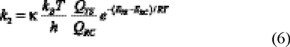
where κ is the tunneling factor which is calculated as the ratio of the quantum-mechanical to the classical barrier crossing rate, assuming an unsymmetrical, one dimensional, Eckart function barrier.
The rate coefficient for each channel overall reaction is obtained by the following expression:
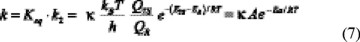
The leucine + OH reaction is a complex system with two different pathways included in the kinetics study; each of them moves forward its own transition structure to a set of various products. To simplify its analysis we have assumed that once a specific pathway started it proceeds to completion, independently of the other pathways, i.e.: there is no mixing or crossover between different pathways. On this basis, the total rate (ktot) constant that measures the rate of the OH disappearance, can be determined by summing up the rate coefficients calculated for two pathways (ktot = kα+kγ) [35]. In addition, in this paper the temperature dependence of k was also studied and calculated Arrhenius parameters are presented.
The tunneling factor, the rate coefficients and the gamma branching ratio (Γγ=kγ /ktot × 100) for the OH hydrogen abstraction reaction from leucine, are reported in Table 2. The rate coefficients for the studied reaction are reported here for the first time. At 298.15 K the proposed values are 1.00 × 108, 1.99 × 109, and 2.09 × 109 L⋅mol-1⋅s-1, for the alpha and gamma channels and the overall reaction, respectively.
Two features of the barrier determine the extent of the tunneling factor: the energy barrier and the barrier width, which is directly related to the imaginary frequency (γ) associated to the transition vector. In the studied reaction no tunneling effect was found because of the relative low barriers and the low frequencies obtained at B3LYP. Compared to other levels of theory , B3LYP usually predicts much lower imaginary frequencies for radical-molecule abstraction reactions.
The gamma rate coefficient was found to be remarkably larger than that of the alpha channel over the all temperature range 250-350 K. The gamma branching ratio slightly decreases with temperature, going from 97% at 250 K to 93% at 350 K. According to kα and kγ values, it seems that in the gas phase the hydrogen abstraction mainly occurs from the gamma site. Nevertheless, the branching ratios can be modified by the peptide environment or by the solvent. This predominance of the gamma channel can be explained by the lower energy barrier, caused by the stronger attractive interaction in the transition state, between the H atom in the OH radical and the N atom in leucine, compared to the alpha TS.
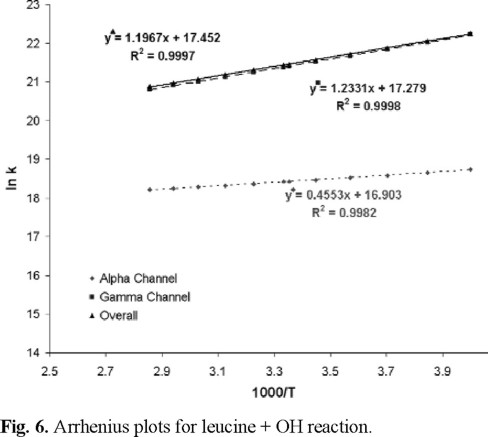
Arrhenius plots are shown in figure 6. The two parameters equation for the overall rate coefficient is ktot = (3.80 ± 0.08) × 107 exp[(2378 ± 12)/T] L⋅mol-1⋅s-1. This expression shows substantial temperature dependence, with negative net activation energy. In addition, the best fits for the alpha and gamma H-abstraction channels were found to be kα = (2.19 ± 0.05) × 107 exp[(905 ± 12)/T], and kγ = (3.19 ± 0.05) × 107 exp[(2450 ± 10)/T] L⋅mol-1⋅s-1, respectively. According to these results, the values of kα, kγ and ktot decrease with temperature. The Arrhenius activation energy for the gamma abstraction is about 2 kcal/mol lower than the one for the alpha abstraction. Apparent activation energy of about -2.4 kcal/mol can be expected for the overall reaction. In all the above expressions the error limits represent 2 standard deviations from the least-squares analysis.
Conclusions
Two competitive channels for the OH hydrogen abstraction reaction from leucine have been considered, corresponding to abstraction from the alpha and gamma sites. The studied paths have been well described as complex reactions. Each of them characterized by a barrier-less first step leading to the formation of a pre-reactive complex, followed by a second step, yielding the corresponding radical and water.
Both optimized transition state geometries show ring-like structures due to the formation of intra-molecular hydrogen bond-like interactions. However, they differ in the atoms involved in the ring. These attractive interactions stabilize the transition structures and reduce the energy barriers. The strongest interaction was found for the gamma H-abstraction channel, and this feature was found responsible of the highest reactivity of this site.
Rate coefficients for the leucine + OH gas phase reaction are reported for the first time and, according to the obtained values, it seems that the hydrogen abstraction occurs almost exclusively from the gamma site.
The temperature dependence of the overall rate constant in L⋅mol-1⋅s-1 can be described by the expression ktot = (3.80 ± 0.08) x 107 exp[(2378 ± 12)/T] and an apparent activation energy of about -2.4 kcal/mol can be expected. In the same units, the rate constants for the alpha and gamma H-abstraction channels are kα = (2.19 ± 0.05) x 107 exp[(905 ± 12)/T] and kγ = (3.19 ± 0.05) x 107 exp[(2450 ± 10)/T] L⋅mol-1⋅s-1, respectively.
Acknowledgements
The authors gratefully acknowledge the financial support from the Instituto Mexicano del Petróleo (IMP) through program D00179. We also thank the IMP Computing Center for super-computer time on SGI Origin 2000. We thank to Professors W. T. Duncan, R. L. Bell and T. N. Truong for providing The Rate program.
References
1. Voet, D.; Voet., J. G. Biochemistry. New York. John Wiley and Sons, Inc., 1990. [ Links ]
2. Stadtman, E. R. Annu. Rev. Biochem. 1993, 62, 797-821. [ Links ]
3. Sies, H. Oxygen Stress, Academic Press: London, 1985. [ Links ]
4. Simic, M. G.; Taylor, K. A.; Ward, J. F.; Von Sonntag, C. Oxygen Radicals in Biology and Medicine. Plenum Press: New York, 1991. [ Links ]
5. Davies, K. J. A. Oxydative Damage and Repair: Chemical, Biological and Medical Aspects. Pergamon Press: New York, 1991. [ Links ]
6. Sies, H. Oxygen Stress-Oxidants and Anti-Oxidants, Academic Press: London, 1991. [ Links ]
7. Von Sonntag, C. The Chemical Basis of Radiation Biology. Taylor & Francis, London, 1987. [ Links ]
8. Beckwith, A. L. J. Tetrahedron 1981, 37, 3073-3100. [ Links ]
9. Giese, B. Radicals in Organic Synthesis; Pergamon Press: Oxford, 1986. [ Links ]
10. Curran, D. P. Synthesis 1988, part 1, 417-439. [ Links ]
11. Goshe, M. B.; Chen, Y. H.; Anderson, V. E. Biochemistry 2000, 39, 1761-1770. [ Links ]
12. Nukuna, B. N.; Goshe, M. B.; Anderson, V. E. J. Am. Chem. Soc. 2001, 123, 1208-1214. [ Links ]
13. Galano, A.; Alvarez-Idaboy, J. R.; Montero, L. A.; Vivier-Bunge, A. J. Comp. Chem. 2001, 22, 1138-1153. [ Links ]
14. Galano, A.; Alvarez-Idaboy, J. R.; Bravo-Perez, G.; Ruiz-Santoyo, Ma. E. J. Mol. Struct. (Theochem) 2002, 617, 77-86. [ Links ]
15. Galano, A.; Alvarez-Idaboy, J. R.; Cruz-Torres, A.; Ruiz-Santoyo, Ma. E. Int. J. Chem. Kinet. 2003, 35, 212-221. [ Links ]
16. Galano, A.; Alvarez-Idaboy, J. R.; Cruz-Torres, A.; Ruiz-Santoyo, Ma. E.; J. Mol. Struct. (Theochem) 2003, 629, 165-174. [ Links ]
17. Alvarez-Idaboy, J. R.; Mora-Diez, N.; Vivier-Bunge, A.; J. Am. Chem. Soc. 2000, 122, 3715-3720. [ Links ]
18. Gaussian 98, Revision A.3, Frisch, M.J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery Jr., J. A.; Stratmann, R. E.; Burant J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian Inc., Pittsburgh PA. 1998. [ Links ]
19. Mayer, I. Int. J. Quantum Chem. 1983, 23, 341-363. [ Links ]
20. Mayer, I. J. Phys. Chem. 1996, 100; 6332-6335. [ Links ]
21. Vargas, R.; Garza, J.; Dixon, D. A.; Hay, B. P. J. Am. Chem. Soc. 2000, 122, 4750-4755. [ Links ]
22. Jansen, H. B.; Ros, P. Chem. Phys. Lett. 1969, 3, 140-143. [ Links ]
23 Boys, S. F.; Bernardi, F. Mol. Phys. 1970, 19, 553-556. [ Links ]
24. Frisch, M. J.; Del Bene, J. E.; Binkley, J. S.; Schaefer III, H. F. J. Chem. Phys. 1986, 84, 2279-2289. [ Links ]
25. Schwenke, D. W.; Truhlar, D. G. J. Chem. Phys. 1985, 82, 2418-2426. [ Links ]
26. Eyring, H. J. Chem. Phys. 1935, 3, 107-115. [ Links ]
27. Evans, M. G.; Polanyi, M. Trans. Faraday Soc. 1935, 31, 875-880. [ Links ]
28. Duncan, W. T.; Bell, R. L.; Truong, T. N. J. Comp Chem. 1998, 19, 1039-1052. [ Links ]
29. Bader, R. F. W. Atoms in Molecules-A Quantum Theory. Oxford University Press, Oxford, 1990. [ Links ]
30. Hammond, G. S. J. Am. Chem. Soc. 1955, 77, 334-338. [ Links ]
31 Rayez, M. T.; Rayez, J. C.; Sawerysyn, J. P. J. Phys. Chem. 1994, 98, 11342-11352. [ Links ]
32. Talhaoui, A.; Louis, F.; Devolder, P.; Meriaux, B.; Sawerysyn, J. P.; Rayez, M. T.; Rayez, J. C. J. Phys. Chem. 1996, 100, 13531-13538. [ Links ]
33. NIST Chemical Kinetics Database on the Web. Public Beta Release 1.1. Standard Reference Database 17, Version 7.0 (http://kinetics.nist.gov/index.php). [ Links ]
34. Singleton D. L.; Cvetanovic, R. J. J. Am. Chem. Soc. 1976, 98, 6812-6819. [ Links ]
35. Robinson, P. J.; Holbrook, K. A. Unimolecular Reactions. Wiley-Interscience: London, 1972. [ Links ]

