











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkANTECEDENTES
El síndrome de Proteus es una enfermedad congénita rara, de carácter hamartomatoso, que puede afectar todas las estructuras del organismo. Este trastorno no se hereda y no existe predilección sexual ni racial. Su nombre se origina del mítico dios griego Proteus, también llamado “el polimorfo” o “el viejo hombre del mar”, quien usaba sus cambios de forma para huir de los que le perseguían para tomar sus habilidades proféticas.1
La referencia más temprana en la bibliografía es la de Graetz, en 1928,2 a propósito de un paciente con hipertrofia corporal. El síndrome de Proteus fue descrito por primera vez en 1979 por Cohen y Hayden; sin embargo, no recibió la denominación hasta 1983 en un artículo publicado por Wiedemann y su grupo, para describir el caso de cuatro niños con malformaciones hamartomatosas congénitas.1 La incidencia estimada es de 1 caso por cada millón de nacidos vivos.3
El síndrome de Proteus es una enfermedad rara, por lo que los pacientes deben recibir asesoría genética, si está embarazada o planea estarlo, si tiene un hijo con un trastorno o defecto congénito, tuvo dos o más pérdidas gestacionales o un hijo que falleció, o si se realizó un ultrasonido o pruebas que sugieren la posibilidad de un problema hereditario.4
Este trastorno genético es esporádico, quizá originado por la función de un gen letal autosómico dominante, que sobrevive por mosaicismo somático.5 Es una alteración genética rara, causada por una mutación somática que activa el gen AKT1, implicado en la vía celular AKT1/ mTOR, como causa importante de la enfermedad.6 Se produce por mutación del gen 10q23.31, encargado de activar de forma esporádica el crecimiento de varios tejidos (epidérmico, conectivo, óseo, adiposo, endotelial) durante el desarrollo embrionario, por lo que se manifiesta al nacimiento o en los primeros años de vida, y se caracteriza por gigantismo parcial de manos y pies, nevos pigmentados, hemihipertrofia cutánea, tumores hamartomatosos subcutáneos, macrocefalia y anomalías craneales.7,8
Suele manifestarse entre los 6 y 18 meses de vida, con crecimiento segmentario progresivo que, con el paso de los meses, se expresa con asimetría importante de las extremidades y el tronco, alcanzando la estabilización luego de la adolescencia.6
Hasta la fecha no existen reportes de madres con síndrome de Proteus y su evolución durante el embarazo, ni sus implicaciones reproductivas.
CASO CLÍNICO
Paciente de 21 años, primigesta, de 88 kg, talla de 1.58 cm, IMC: 35.4, sin antecedentes heredofamiliares de importancia. La paciente refirió manifestaciones de síndrome de Proteus desde el nacimiento; durante la niñez se le practicó la exéresis de un lipoma lumbar, sin complicaciones aparentes; a los 6 años se estableció el diagnóstico, por menarquia temprana, pubarquia a los 10 años. Recibió tratamiento con inhibidores de la hormona de crecimiento y permaneció en seguimiento por los especialistas del servicio de Genética pediátrica, Reumatología, Psicología, Psiquiatría, Ortopedia y Oftalmología. Antes de los 15 años se le diagnosticó asma. El oftalmólogo estableció el diagnóstico de miopía y la trató con el uso correctivo de lentes. En la adolescencia manifestó alteraciones vasculares, y el angiólogo le diagnosticó insuficiencia venosa periférica. Desde entonces permanece en tratamiento con medias de mediana compresión y ácido acetilsalicílico. Inició su vida sexual a los 18 años, y no ha tenido detecciones oportunas de cáncer cervicouterino.
Acudió a control prenatal al servicio de Medicina Materno Fetal del Hospital General de México Dr. Eduardo Liceaga, a las 18 semanas de embarazo, sin complicaciones. Refirió consumo de fármacos hematínicos y multivitamínicos desde la confirmación del embarazo. Cursó con infección de vías urinarias en el primer trimestre, con tratamiento médico no especificado y mejoría del cuadro clínico; también se diagnosticó condilomatosis vulvar, sin tratamiento.
Se otorgó tratamiento multidisciplinario por el servicio de Genética y Dermatología. Durante todo el embarazo recibió 100 mg de ácido acetilsalicílico cada 24 h, dosis profiláctica hasta su interrupción, debido al riesgo de trombosis por las alteraciones vasculares y su enfermedad de base y, per se, por el embarazo. Se recomendó la interrupción del embarazo por vía abdominal, con seguimiento en la consulta posterior a la finalización de la cesárea debido a várices vulvares, con uso de medias de mediana compresión y toma de ácido acetilsalicílico.
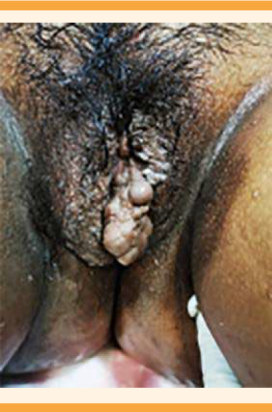
El padecimiento se inició a las 35 semanas de embarazo, por fecha de la última menstruación, después de acudir a la consulta de control prenatal, donde se le diagnosticó elevación de las cifras tensionales. A su llegada al área de triage de Ginecología reportó: tensión arterial de 155-101 mmHg, posteriormente corroborada de 175-123 mmHg, y cefalea. En el área de admisión se inició tratamiento activo para preeclampsia. La hemodinamia materna reportó un gasto cardiaco de 4 L/min, índice cardiaco 2 L/min, resistencias vasculares sistémicas de 2835 dynas, resistencias vasculares sistémicas indexadas 5775 dynas. Se le administró reanimación hídrica inicial con soluciones cristaloides Hartmann 300 cc en carga de dosis única; posteriormente, 1 bolo de labetalol de 5 mg y nifedipino 30 mg. Se prescribió un esquema de Zuspan de impregnación, con 4 gramos de sulfato de magnesio porque refirió cefalea; a los 10 minutos se corroboró la tensión arterial que se encontró en 155-108 mmHg, por lo que se administró la segunda dosis de 5 mg de labetalol por vía intravenosa, con adecuada reacción a los 10 minutos, con tensión arterial de 148-92 mmHg. Se le diagnosticó preeclampsia, conforme a los criterios de severidad por cifras tensionales y síntomas. Los estudios de laboratorio reportaron: leucocitos 7.30 x 103/µL, hemoglobina 11.8 g/dL, plaquetas 145 x 103/µL, glucosa 67 mg/dL, creatinina 0.53 mg/dL, ácido úrico 4.7 mg/dL, bilirrubina directa 0.02 mg/dL, bilirrubina indirecta 0.22 mg/dL, TGP 7 U/L, TGO 12 U/L, examen general de orina con proteínas 600 mg/dL (Cuadro 1). A la exploración física se observaron: hipertrofia bilateral de manos, de predominio izquierdo (Figura 1) e hipertrofia de la pierna derecha (Figura 2); fondo uterino de 30 cm, a expensas de útero gestante. El peso estimado del feto, por ultrasonido, fue de 2193 g, con 33.3 semanas de embarazo por fetometría, hemodinamia fetal con arteria cerebral media IP 1.18 en percentil 3 (patológico), índice cerebro-placentario IP 0.84 en percentil menor de 1 (patológico); todos los trayectos medidos con diástole anterógrada. No se observó ningún saco de líquido amniótico medible. En los genitales (labios mayores) se observaron condilomas menores de 1 cm y várices vulvares (Figura 3), por lo que se decidió la interrupción del embarazo por vía abdominal, por diagnóstico de anhidramnios y pérdida del bienestar fetal. Nació una mujer de 2350 g (percentil 32), talla de 45 cm, Apgar 8 y 9, Capurro 35.2, Silverman 3, sangrado total estimado de 1000 cc. El nuevo estudio de hemodinamia materna reportó valores dentro de los parámetros normales. No se advirtieron complicaciones; la paciente permaneció con cifras tensionales arteriales medias menores a 106 mmHg, con 100 mg de metoprolol cada 12 h, 60 mg de nifedipino cada 12 h y 40 mg de enoxaparina por vía subcutánea en dosis profilácticas. Cursó con buena evolución clínica, con estudios de laboratorio de control en parámetros normales (Cuadro 1). Se decidió el alta del servicio a las 48 h, por mejoría clínica. Siete días después se retiraron los puntos de sutura quirúrgica sin eventualidades y, progresivamente, se retiró el medicamento hipertensivo por hipotensión. En la actualidad, la paciente permanece en seguimiento en los servicios de Dermatología, Genética y Vascular periférico por várices. Se cambió el esquema de tratamiento, de enoxaparina a aspirina 100 mg por vía oral cada 24 h de mantenimiento, para continuar en vigilancia médica. La recién nacida permaneció en observación, en el cunero patológico durante 48 h debido a taquipnea transitoria, por lo que recibió oxígeno complementario, con reacción satisfactoria. La madre y su hija fueron dadas de alta del hospital sin complicaciones aparentes.
Cuadro 1 Evolución de los estudios de laboratorio al ingreso y egreso
| Estudios de laboratorios | 31/12/2019 (al ingreso) | 02/01/2020 (al egreso) |
|---|---|---|
| Leucocitos (x10e3/uL) | 7.3 | 9.4 |
| Neutrófilos (%) | 56.1 | 90.4 |
| Hemoglobina (gr/dL) | 11.8 | 9.0 |
| Hematocrito (%) | 34.5 | 28.6 |
| Plaquetas (x10e3/uL) | 145 | 130 |
| Glucosa (mg/dL) | 67 | 142 |
| Urea (mg/dL) | 24.1 | 43.3 |
| Creatinina (mg/dL) | 0.53 | 0.84 |
| Ácido úrico (mg/dL) | 4.7 | 6.2 |
| BT (mg/dL) | 0.24 | 0.22 |
| BD (mg/dL) | 0.02 | 0.04 |
| BI (mg/dL) | 0.22 | 0.18 |
| Albúmina (g/dL) | 2.29 | 2.68 |
| Proteínas (g/dL) | 5.4 | 4.8 |
| TGO (U/L) | 12 | 15 |
| TGP (U/L) | 7 | 8 |
| DHL (U/L) | 174 | - |
| Examen general de orina | pH 6.5, leucocitos 4 x campo, eritrocitos 1 x campo, proteínas 600 mg/dL. | pH 5.5, leucocitos 1-3 x campo, eritrocitos 3-5 x campo, proteínas 70 mg/dL. |


DISCUSIÓN
Desde la primera descripción y hasta la fecha, se han diagnosticado poco más de 500 casos de síndrome de Proteus en países industrializados. Al respecto, parece haber una tendencia de afectación a varones. Constituye una enfermedad progresiva que implica desfiguraciones severas, limitaciones físicas y reducción de la calidad de vida.9,10 En la paciente del caso, la expresión en mosaico y el tratamiento multidisciplinario desde la infancia superaron la esperanza de vida.
El análisis cromosómico de estos pacientes es común. Solo en 1 caso se detectó un segmento adicional en el brazo largo del cromosoma 1, que sugiere una mutación somática.11,12 El gen no es trasmisible porque cuando se expresa en el cigoto induce la muerte temprana del embrión.13,14 En pacientes con este tipo de alteraciones hereditarias se sospecha que la enfermedad puede ocasionar pérdida gestacional recurrente.
En la paciente del caso, y en la mayoría de los casos descritos, no se observó el cuadro clínico completo. El diagnóstico requiere una alta sospecha clínica.6 Las manifestaciones son más evidentes en los primeros años de vida y alcanzan la estabilización luego de la adolescencia.
El estrabismo, las cataratas y las convulsiones suelen afectar de forma importante la calidad de vida.3 Entre las manifestaciones clínicas del síndrome de Proteus se encuentran:1
Hemihipertrofia corporal: suele ser completa, pero se han reportado algunos casos de tipo parcial. No existe predilección por el lado derecho o izquierdo.
Gigantismo parcial de manos y pies: de manera bilateral, con hipertrofia de tejidos blandos. En nuestro caso, la paciente tuvo hipertrofia bilateral de manos, con predominio izquierdo (Figura 1), y de la pierna derecha (Figura 2).
Tumores de partes blandas: son de rápido crecimiento y aparecen en el tronco o la parte proximal de los miembros. Los más frecuentes son: lipomas, hamartomas, hemangiomas cavernosos, linfangiohemangioma. La paciente tenía antecedente de lipoma con exéresis durante la infancia.
Alteraciones óseas: casi siempre en el cráneo y se observan como protuberancias en la zona fronto-temporal.
Alteraciones oculares: exostosis periorbitarias, iris heterocromático, coloboma retiniano, nistagmo, estrabismo, escleróticas azules, telecantus, epibléfaron y hemimegalia del nervio óptico. 15
Alteraciones genitourinarias: quistes renales y epididimales.18
Crecimiento acelerado asimétrico: suele expresarse en los primeros años de vida, aunque la talla adulta final suele ser normal. Los valores analíticos de hormonas tiroideas y hormona del crecimiento no se alteran.
Neuropatías: desmielinización, compresión del cuerpo calloso, hemimegaloencefalia, nódulos calcificados subependimales, quistes periventriculares, craneosinostosis, epilepsia, retraso mental.19,20
Lesiones cutáneas: sobre todo de tipo pigmentario, pero pueden observase:
Se han propuesto diversos criterios diagnósticos, los primeros fueron los de Wiedemann y su grupo.21 Con el paso del tiempo, diversos autores han descrito diferentes signos clínicos: los de Clark en 1987,22 Costa en 198523, Samlaska en 198924 y Hotamisligil en 199025 (Cuadro 2). En la actualidad, el diagnóstico se establece con los hallazgos clínicos, radiológicos y evolutivos. El signo más específico es la coexistencia de una superficie plantar de aspecto cerebriforme. 26
Cuadro 2 Signos clínicos del síndrome de Proteus, según los criterios de diferentes autoresw
| Clark, et al (1987)22 y Costa, et al (1985)23 | Samlaska, et al (1989)24 | Hotamilligil (1990)25 | Institutos Nacionales de Salud, EUA (1998)27 |
|---|---|---|---|
|
Principales
Hemihipertrofia corporal Megadactilia Nevos epidérmicos Tumores subcutáneos Engrosamiento de la dermis Crecimiento acelerado Frecuentes Macrocefalia Amiotrofia Tumores profundos Retraso psicomotor Estrabismo Malformación craneal Dilataciones venosas Ocasionales Máculas acrómicas Manchas café con leche Convulsiones Malformaciones cerebrales Macroftalmia Ptosis Cataratas Miopía Nistagmo |
Criterios
mayores
Hemihipertrofia (parcial o completa) Macrodactilia Masas subcutáneas Lipomatosis Lipomatosis pélvica Hamartomas Hemangiomas cavernosos Linfangiohemangiomas Linfohemangioma Lipohemangioma Hamartoma hemolinfoangiomatoso Masas plantares o palmares Nevos del tejido conectivo Lipomatosis Exostosis craneales o en extremidades distales Nevos epidérmicos lineales o espirales Escoliosis Criterios menores Cutáneos: varicosidades, prominencias venosas, despigmentaciones, manchas “café con leche”,angioqueratomas, nevos del tejido conectivo, ausencia de mamas, neurofibroma, fibromas, disminución del tejido subcutáneo Esqueléticos: macrocefalia, crecimiento aumentado, genu valgus, cuello largo, anquilosis de codo, cifosis, pectum excavatum, retardo de crecimiento óseo, pies anchos, dislocación de la cadera Craneofaciales: estrabismo, defectos oculares, mala oclusión dentaria, paladar arqueado, pequeña circunferencia occipitofrontal, prognatismo mandibular, puente nasal disminuido, bajo asiento de los pabellones auriculares Otros: retaso mental, atrofia muscular, abdomen prominente, anomalías quísticas pulmonares, crisis convulsivas, desarrollo precoz mamario, piernas musculosas, nódulos en las cuerdas vocales, hipertrofia de pene, nariz picuda |
Macrodactilia y
hemihipertrofia: 5 puntos Engrosamiento palmar o plantar: 4 puntos Tumores subcutáneos y lipomas: 4 puntos Nevos epidérmicos: 3 puntos Macrocefalia o exostosis craneales: 2.5 puntos Al resto de anormalidades ´menores: 1 punto |
A
1. Nevos del tejido conectivo B 1. Nevos epidérmicos 2. Crecimiento desproporcionado (uno o más) de los miembros: Brazos y piernas Manos, pies y dedos Cráneo Hiperostosis Conducto auditivo externo Hiperostosis Vértebras Megaespondilodisplasia Vísceras Bazo y timo 3. Tumores específicos antes de finalizar la segunda década (cualquiera de ambos) Cistadenomas bilaterales de ovario Adenoma monomorfo de parótida C 1. Tejido adiposo no regulado (solo uno) Lipomas Ausencia regional de grasa 2. Malformaciones vasculares (una o más) Malformaciones capilares Malformaciones venosas Malformaciones linfáticas 3. Fenotipo facial Dolicocefalia Cara alargada Inclinación leve debajo de las comisuras palpebrales |
| Si el paciente tiene cuatro de los siete criterios se establece el diagnóstico de la enfermedad. | Si el paciente cumple con cuatro de los siete criterios mayores se establece el diagnóstico de la enfermedad. | El diagnóstico se establece si el paciente suma 13 o más puntos | El único criterio específico incluido en la categoría A, ante todos los criterios obligatorios es suficiente para establecer el diagnóstico, además de dos criterios de la categoría B o tres de la C. |
Para clarificar estos criterios, en marzo de 1998 se convocó la primera Conferencia Nacional del Síndrome de Proteus para Padres y Familiares (National Institutes of Health de Bethesda, Maryland, EUA), 27 donde se establecieron una serie de criterios diagnósticos, recomendaciones, guías de evaluación y diagnósticos diferenciales. Los criterios generales (obligatorios) fueron: distribución de las lesiones en mosaico, curso progresivo y aparición esporádica; por su parte, los criterios específicos se clasificaron en las categorías A, B y C. El criterio incluido en la categoría A: la coexistencia de todos los criterios obligatorios es suficiente para establecer el diagnóstico, así como dos de la categoría B o tres de la C.27
Las pruebas diagnósticas deben incluir:
Tomografía computada: útil para evaluar las malformaciones quísticas pulmonares.
Resonancia magnética abdominal: importante para identificar lipomas ocultos, que pueden tener un comportamiento agresivo.
Resonancia magnética del sistema nervioso central: imprescindible para descartar anomalías adicionales.26
Cariotipo: debido a la posibilidad de encontrar alguna translocación o pérdida.27
Radiografías óseas seriadas.
Entre los diagnósticos diferenciales es necesario destacar otro tipo de enfermedades hamartomatosas: neurofibromatosis, síndromes de Kipplel-Trenaunay, Mafucci, Bannayan-Riley-Ruvalcaba, Parkes-Weber, nevo epidérmico, lipomatosis simétrica y lipomatosis encefalocraneocutánea.1,28
El pronóstico se establece según la ubicación y el grado de sobrecrecimiento de los tejidos afectados, además de otras complicaciones importantes: bulas pulmonares y anormalidades del sistema nervioso central.6 La principal causa de muerte prematura es la tromboembolia pulmonar y la insuficiencia respiratoria aguda; la mortalidad estimada es de 25% antes de los 20 años.29 En el caso aquí expuesto, la tromboprofilaxis mecánica y el ácido acetilsalicílico desde la adolescencia y durante el embarazo demostraron su eficacia como tratamiento.
El tratamiento del síndrome de Proteus debe ser multidisciplinario, con la intervención de un dermatólogo, genetista, traumatólogo, vascular periférico, nutricionista, oftalmólogo, cirujano, neurólogo y salud mental.30
Existen nuevas terapias relacionadas con la vía PI3K/AKT/mTOR. El ARQ 092 es un fármaco que se administra por vía oral, es un inhibidor alostérico selectivo del AKT, evaluado in vitro en células de pacientes con síndrome de Proteus con mutaciones activantes del gen AKT1, que ha demostrado disminuir la fosforilación del AKT e inhibir la cascada de señalización. En la actualidad están en marcha ensayos clínicos con ARQ 092 en pacientes con síndrome de Proteus, cuyo objetivo es mejorar la calidad de vida y reducir los síntomas.31
CONCLUSIÓN
Hasta la fecha siguen sin conocerse las implicaciones en la salud sexual y reproductiva de las mujeres con síndrome de Proteus; el informe de este caso fue exitoso. Las malformaciones vasculares y las alteraciones del tejido adiposo sugieren un factor de riesgo que altera la regulación de la angiogénesis y la insuficiencia venosa supone un promotor de trombosis; a su vez, esto puede considerarse riesgo de preeclampsia, por lo que se sugiere el tratamiento multidisciplinario temprano para evitar complicaciones. Per se, deben informarse y recopilarse más casos para aceptar esta hipótesis.

