










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista mexicana de cardiología
versión impresa ISSN 0188-2198
Rev. Mex. Cardiol vol.26 no.4 México oct./dic. 2015
Original Research
The adult with congenital heart disease
El adulto con cardiopatía congénita
Lucelli Yáñez-Gutiérrez,* Diana López-Gallegos,* Carmen Emma Cerrud-Sánchez,* Horacio Márquez-González,* Marlenne Berenice García-Pacheco,* Moisés Jiménez-Santos,** Jaime Alfonso Santiago-Hernández,*** Homero Alberto Ramírez-Reyes,*** Carlos Riera-Kinkel****
* Servicio de Cardiopatías Congénitas.
** Servicio de Tomografía.
*** Servicio de Hemodinamia.
**** División de Cirugía Cardiotorácica.
UMAE Hospital de Cardiología Centro Médico Nacional Siglo XXI.
Correspondence to:
Lucelli Yáñez Gutiérrez MD
Cuauhtémoc Núm. 330, Col. Doctores,
Del. Cuauhtémoc, 06760, Mexico City
Phone: (55) 56-27-69-00, Local: 22203
E-mail: lucelli.yanez@imss.gob.mx
ABSTRACT
Background: The study show the most important demographic and clinical data of a cohort composed by adults treated in a congenital heart clinic. Material and methods: In a retrospective, observational and longitudinal study, data from patients with congenital heart disease, older than 18 years, seen between May 2010 and May 2013, were analyzed. Results: The cohort comprised 409 patients, 280 of them female (69%), whose mean age at the time of the first evaluation was 36.7 ± 14.2 years (range, 18 to 75 years). Thirty-four of them (8%) had cyanotic congenital heart disease (14 cases of tetralogy of Fallot, the rest with a univentricular heart and Eisenmenger). The mean age when heart disease was diagnosed was 33.6 ± 15.9 years (range, from birth to 75 years). One hundred five patients (38%) were aware of having heart disease but did not have regular follow-up. The reason for referral was heart murmur in 143 subjects (35%); in 143 (35%) for deterioration of functional class due to progressive dyspnea; supraventricular arrhythmias in 24 (6%); chest pain in 16 (4%), in 12 arterial hypertension (3%) and in 8 (2%) history of cerebral vascular event. On regard of the types of congenital heart disease, 71% were intracardiac defects (mainly interatrial and interventricular septal defects), 10% left and right obstructive lesions, as well as aortic coarctation, 7% mixed valve lesions, 2% cardiomyopathy, and the rest with diseases like anomalous origin of the left coronary artery, double discordance and univentricular heart. In 188 patients (46%) surgical treatment was opted, meanwhile in other 138 (34%) interventional percutaneous treatments were chosen, and in 83 (20%) medical treatment was selected. Fifteen patients suffered complications related to treatment, 50% of them infections, arrhythmias or heart failure. Mortality occurred in six cases, all from the surgical group. The cause of death was refractory heart failure and cardiogenic shock in the postoperative phase. Pulmonary pressure descended from 48.3 ± 20.4 mmHg before the therapeutic procedure to 36.3 ± 16.2 mmHg after it (p < 0.001). Conclusions: Adults with congenital heart disease form a complex study group that in addition to the underlying disease, have a combination of other diseases and/or cardiovascular risk factors as well as several complications related to previous therapeutic procedures.
Key words: Congenital heart disease, adults, complications of treatment, follow-up of congenital heart diseases.
Antecedentes: Este estudio muestra los datos clínicos y demográficos más importantes de una cohorte compuesta de pacientes tratados en una clínica de cardiopatías congénitas. Material y métodos: Fueron analizados los datos de pacientes con cardiopatías congénitas, mayores de 18 años, vistos entre los meses de mayo del 2010 al 2013, en un estudio retrospectivo, observacional y longitudinal. Resultados: La cohorte comprendió 409 pacientes, 280 de ellos del género femenino (69%), con promedio de edad de 36.7 ± 14.2 años (rango de 18 a 75 años). Treinta y cuatro de ellos (8%) tenían cardiopatía congénita cianótica (14 con tetralogía de Fallot, el resto con corazón univentricular o Eisenmenger). El promedio de edad en la que fue diagnosticada la cardiopatía fue de 33.6 ± 15.9 años (rango, desde el nacimiento a los 75 años). Ciento cinco pacientes (38%) conocían que tenían cardiopatía, pero no tenían un seguimiento regular. La razón de la referencia fue en 143 individuos (35%) un soplo cardiaco, en otros 143 (35%) por deterioro de la clase funcional debido a disnea progresiva; arritmias supraventriculares en 24 (6%); dolor torácico en 16 (4%), hipertensión arterial en 12 (3%) y en 8 (2%) historia de evento vascular cerebral. En cuanto al tipo de cardiopatías congénitas en 71% de los casos fueron defectos intracardiacos (principalmente, comunicaciones interauriculares e interventriculares), 10% de lesiones obstructivas derechas e izquierda, así como coartación de la aorta; 7% de lesiones valvulares combinadas; 2% de cardiomiopatías y el resto que comprendió enfermedades como nacimiento anómalo de la coronaria izquierda, doble discordancia y corazón univentricular. En 188 pacientes (46%) los pacientes fueron tratados quirúrgicamente, en tanto que en 138 (34%) se decidió por la intervención percutánea y en 83 (20%) más, por el tratamiento médico. Quince pacientes tuvieron complicaciones relacionadas con el tratamiento, 50% de ellas infecciones, arritmias o insuficiencia cardiaca. Seis pacientes del grupo quirúrgico murieron, debido a insuficiencia cardiaca refractaria o choque cardiogénico en el postoperatorio. La presión de la arteria antes y después del procedimiento terapéutico bajó de 48.3 ± 20.4 a 36.3 ± 16.2 mmHg (p < 0.001). Conclusiones: Los adultos con cardiopatía congénita forman un grupo de estudio complejo, pues en ellos se agregan a la enfermedad de base otras enfermedades y factores de riesgo cardiovascular, más las complicaciones relacionadas con los procedimientos terapéuticos previos.
Palabras clave: Cardiopatía congénita, adultos, complicaciones del tratamiento, seguimiento de las cardiopatías congénitas.
INTRODUCTION
Pediatric cardiology is a relatively young and booming branch of cardiovascular medicine with exponential growth in recent years. Due to advances in surgical techniques, improved devices used in interventional cardiology and better postoperative care of patients, survival rates in patients with congenital heart disease (CHD) have increasead.1 It is estimated that a great proportion of children with CHD reach adulthood, to the point that, for the first time, the number of adults with congenital cardiopathies exceeds the amount of children with the same pathology.2,3
Only in the United States there are about one million adult patients with CHD, with an annual growth rate of 5%. In that country, every year 50,000 more of these patients will require attention. In our country, we can estimate 300,000 cases of adults with CHD, with an annual increase of 15%.4
The largest group of adult patients with CHD is composed by those patients who underwent some kind of therapeutic intervention during childhood. This is the reason why we are now detecting more late complications of these interventions. Reasons for referral in this group of patients are mainly heart failure, endocarditis, diverse arrhythmias and pregnancy.5
The scope of this study is to assess and analyze some clinical data in a cohort of adult patients with CHD followed-up in the outpatient clinic of our service.
MATERIAL AND METHODS
The present study is retrospective, observational and longitudinal. Data from clinical records of patients older than 18 years, treated in the service of congenital heart disease of our service, in the period from May 2010 to May 2013 were analyzed. In addition to demographic data, we took into consideration the type of CHD, the reason of referral to our service and the kind of therapeutic strategy employed: surgical, interventional or medical.
Descriptive basic statistic methods were employed to measure central tendency and dispersion, according to distribution and inferential statistics.
RESULTS
The clinical data of 409 patients were analyzed, 280 of them were female (69%). The age at the time of evaluation was 36.7 ± 14.2 years (range 18-75 years).
At the time of evaluation, there were only 34 cases of cyanotic CHD (8%); of these, 14 patients had tetralogy of Fallot, 10 univentricular heart and 10 Eisenmenger.
The age at the time when the diagnosis of CHD was established was 33.6 ± 15.9 years on average (ranging from birth to 75 years). Of all patients, 38% patients knew they had heart disease and failed to undergo follow up, mostly because they lost their pertaining to social security.
The main reasons for referral were: heart murmur, deterioration of the New York Heart Association (NYHA) functional class due to dyspnea, arrhythmia, chest pain, systemic arterial hypertension, and presence of cerebral vascular event (Figure 1).
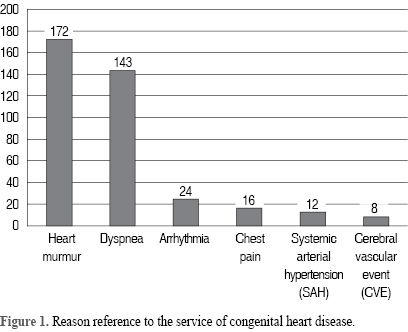
Figure 2 shows the main diagnoses in order of frequency: intracardiac septal defects (290), left side obstructive lesions (41), valve lesions (29), cardiomyopathies (8) and mixed lesions (41).
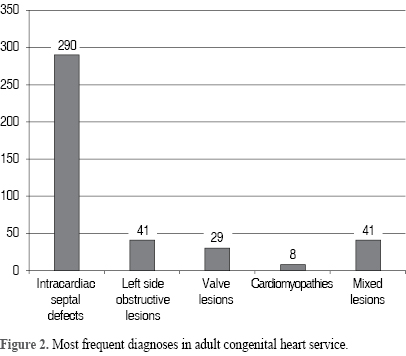
Figure 3 provides a breakdown of the most common diagnoses seen during the observation interval.

Figure 4 shows the type of therapeutic options employed during the observation time. Surgical treatment included: closure of atrial or ventricular septal defects in two cases associated with myocardial revascularization and implantation of mechanical prosthesis. Mechanical prosthesis placement was either for the first time or involved valved tube or valve replacements. Reoperation included interventions to correct residual defects or total reparation and was mostly for tetralogy of Fallot or, double outlet right ventricle including a small group of adult patients who undergo complete Fontan surgery.

The interventional treatment mostly encompassed closing atrial septal defects and patent ductus arteriosus but also included aortoplasty patients with angioplasty and stenting of pulmonary peripheral branches, as well as a small but significant number of cases in which embolization of collateral arteries was performed as a preparation for completing univentricular heart surgery. And finally, in the medical treatment group were included patients with heart failure, pulmonary hypertension or cardiac arrhythmias, whatever this type of treatment was the only alternative or concomitantly with either of the other two options.
Special mention should be made of the group of patients with short circuits, essentially interatrial septal defect with pulmonary artery systolic pressure (PASP > 40 mmHg) as they all underwent cardiac catheterization study with vascular reactivity tests with nitric oxide. Those who showed a decrease in pulmonary artery pressure were considered candidates for a surgical technique with valved patch pulmonary pressure showed a statistically significant decrease of 48.3 ± 20.4 versus 36.3 ± 16.2 (p < 0.001).
In the surgical group occurred six deaths for cardiogenic shock, and four cases for infection, arrhythmias and heart failure.
DISCUSSION AND CONCLUSIONS
Six to eight children are born with heart problems for each 1,000 live births. Half of the children with CHD require urgent attention in the first year of life, while the majority of the rest will require, at some time, medical specialized attention or therapeutic intervention. As it has been mentioned previously, the fact that 8 out of 10 reach patients with CHD will reach adulthood, thanks to advances in diagnostic and therapeutic techniques,1 explains that for the first time the amount of adults with CHD exceeds the number of pediatric patients with these diseases. In consequence an increased number of adult patients with CHD now are seen and studied in cardiology departments, making it necessary to know better the physiopathology and potential complications of these heart problems, as well as the therapeutic options (surgical or interventional) they require.2
A significant number of patients were not aware of their heart disease, while about a third of them indeed had the information that they had received specialized attention, at some point in their lives. In spite of this fact, many of them had not been under regular follow-up, either because they consider the heart problem essentially benign or for administrative reasons, mainly the loss of their right to social security medical care from the moment they reach adulthood. In the group of patients with recent diagnosis of CHD, the most numerous are those with atrial septal defects or patent ductus arteriosus, because, mostly of all, atrial septal defects (and less commonly patent ductus arteriosus) are well tolerated during childhood and adolescence, developing symptoms around the 4th or 5th decade of life (generally the clinical manifestations were the deterioration of functional class due to dyspnea and cardiac arrhythmias, generally of supraventricular origin).6,7
The group of patients undergoing intervention included those with left obstructions either valve or aortic coarctation. Also, tetralogy of Fallot appears as the first complex heart disease treated in adulthood after being operated during childhood. Furthermore, a very small number of patients with univentricular heart syndrome were included.8,9
Similar to what happens in the pediatric age, in adults a very common cause of referral is the detection of a cardiac murmur during a routine examination, but in the latter age this clinical finding is more frequently associated to a decline in functional class due to dyspnea on moderate efforts, as well as the apparition of cardiac arrhythmias.1
Unlike what happens in childhood, in adults with CHD surgery is the first choice for many diseases. But unfortunately, there is a large group of patients in whom surgical procedures are not indicated because the coexistence of heart failure or pulmonary arterial hypertension secondary to voluminous systemic-pulmonary shunts caused by large defects. When pulmonary vessels are severely damaged and are unresponsive to pulmonary vasodilators, in general, curative procedures have to be ruled out. And finally, in a very small number of patients, especially those with complex heart disease, a combination of surgical, interventional and medical procedures could be necessary.7,8
According to the above mentioned, it is necessary to delve in the natural history of these diseases, in the possible therapeutic approaches and their inherent complications, because the initial anatomic and physiological complexities of some CHD are entangled in the adult to other acquired heart diseases, to the effects of pregnancy and to the late complications of the therapeutic procedures. In order to provide the better care to these rather complex patients a multidisciplinary team is mandatory formed by pediatric and adult cardiologists, echocardiographers, interventional, and electrophysiology cardiologists, as well as cardiac surgeons, intensive care specialists, psychologists, geneticist, etc.10-12
The challenge of the future is to raise consciousness in everyone for earlier diagnosis and timely treatment. Also is needed to inculcate in the parents of children with CHD the strict compliance of the following-up strategies, beginning with regular and periodical medical visits. These are diseases of lifetime duration, needing constant supervision and guidance. Finally, is highly desirable the creation of multidisciplinary clinics able to bring high quality care to these patients, as well as constitute themselves in centers for promoting education and research, encouraging effective communications between all elements of the health team.2,13
REFERENCIAS
1. Williams RG, Pearson GD, Barst RJ et al. Report of the National Heart, Lung and Blood Institute Working Group on research in adult congenital heart disease. J Am Coll Cardiol. 2006; 47 (4): 701-707. [ Links ]
2. WrenC, O'Sullivan JJ. Future demand for follow-up of adult survivors of congenital heart disease. Heart. 2001; 85: 438-443. [ Links ]
3. Hoffman JI, Cristianson R. Congenital heart disease in a cohort of 19,502 births with long-germ follow-up. Am J Cardiol. 1978; 42: 641-467. [ Links ]
4. Ruiz O. Congenital heart disease in adults: residua, sequelae, and complications of cardiac defects repaired at an early age Rev Esp Cardiol. 2003; 56: 73-88. [ Links ]
5. Warnes C, Liberthson R, Danielson G et al. Task Force 1: the changing profile of congenial heart disease in adult life. J Am Coll Cardiol. 2001; 37: 1161-1198. [ Links ]
6. Bricker ME, Hillis LD, LAnge RA. Congenital heart disease in adults: first of two parts. N Engl J Med. 2000; 342: 256-263. [ Links ]
7. Perloff JK, Warnes CA. Challenges posed by adults with repaired congenital heart disease. Circulation. 2001; 103: 2637-2643. [ Links ]
8. Gilboa SM, Salemia JL, Nembhard WN et al. Mortality resulting from congenital heart disease among children and adults in the United States, 1999 to 2006. Circulation. 2010; 122: 2254-2263. [ Links ]
9. Khairy P, Ionescu-Ittu R, Mackie AS et al. Changing mortality in congential heart disease. J Am Coll Cardiol. 2010; 56: 1149-1157. [ Links ]
10. Warnes CA, Liberthson R, Danielson GK et al. Task force 1: the changing profile of congenial heart disease in adult life. J Am Coll Cardiol. 2001; 7: 1170-1175. [ Links ]
11. Oliver R. Cardiopatías congénitas del adulto: residuo, secuelas y complicaciones de las cardiopatías congénitas operadas en la infancia. Rev Esp Cardiol. 2003; 56: 73-88. [ Links ]
12. Warnes CA. The adult with congenital heart disease: born to be bad? J Am Coll Cardiol. 2005; 46: 1-8. [ Links ]
13. Bhatt A, Foster E, Kuehl K et al. Congenital heart disease in the older adult. A scientific statement from the American Heart Association. Circulation. 2015; 131: 1884-1931. [ Links ]
Nota
Este artículo puede ser consultado en versión completa en: http://www.medigraphic.com/revmexcardiol

