











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El cáncer colorrectal (CCR) es una enfermedad compleja en cuyo desarrollo intervienen factores genéticos y ambientales, así como la evolución propia de la enfermedad y la respuesta al tratamiento.1 El CCR es el cuarto cáncer más común en todo el mundo y la tercera causa de muerte por cáncer.2 De acuerdo con el modelo de Vogelstein, el desarrollo del CCR se inicia con la adquisición de una mutación “driver” en alguno de los oncogenes o genes supresores de tumores.3
Recientemente se han identificado variantes involucradas en el desarrollo del cáncer en el gen supresor de tumor TSC2, el cual regula el crecimiento y la proliferación celular mediante la inactivación de la vía del fosfatidilinositol-3-cinasa que inhibe al complejo mTOR (mammalian target of rapamycin).4 TSC2 forma un heterodímero con TSC1 y ambos han sido estudiados como reguladores del crecimiento tumoral en pacientes con esclerosis tuberosa, caracterizada por la presencia de tumores benignos en piel, cerebro, riñón, pulmón, retina y corazón.5,6 Se ha descrito una asociación de variantes del complejo TSC1/TSC2 con el desarrollo de cáncer, incluido el CCR.7,8
En este trabajo determinamos las frecuencias alélicas y genotípicas de dos variantes de TSC2 recientemente incluidas en Catalogue of Somatic Mutations In Cancer (COSMIC):
c.3915G>A (COSV51911975), ubicada en el exón 33, involucra un nucleótido no conservado y se ha relacionado con meningioma y CCR.
c.5371G>A (COSV51913362), en la que se reemplaza el aminoácido glicina con un residuo de serina (variante AA = p.G1791S) y cuyo significado es incierto.9,10 Posteriormente exploraremos su posible asociación con el desarrollo de CCR en población mexicana.
Métodos
Muestras y diseño del estudio
Se incluyeron muestras de ADN extraído de sangre periférica de 126 pacientes con diagnóstico de CCR esporádico y 134 de sujetos sanos sin historia familiar de cáncer, obtenidas de un banco de sangre. Se incluyeron pacientes con CCR esporádico, de ambos sexos, con edad de 18 a 80 años, sin metástasis y sin características clínicas de esclerosis tuberosa. Los pacientes fueron captados en el Hospital Alfredo Pumarejo (Matamoros, Tamaulipas, México) durante 2017 y 2018. Las muestras del grupo de control fueron obtenidas del área clínica del Centro Universitario del Sur en Ciudad Guzmán, Jalisco, México.
Extracción del ADN y PCR-RFLP
El ADN se extrajo a partir de 5 mL de sangre periférica mediante el método de Miller.11 La pureza y la cuantificación del ADN se analizaron con un equipo NanoDrop ONE™ (Thermo Fisher Scientific). Las variantes alélicas se identificaron mediante el método PCR-RFLP tradicional. El diseño de los iniciadores utilizados para la amplificación de los fragmentos de las variantes se realizó con el programa Primer-Blast (National Center for Biotechnology Information, National Library of Medicine, Estados Unidos).12 Los fragmentos amplificados fueron de 215 pares de bases en la variante c.3915G>A, para lo cual se usó el iniciador sentido 5’ AGGAGAAGGCTG GTTCTCG 3’ y el iniciador antisentido 5’ GCCTACAGCAGGGTGAGTGT 3’. En la variante c.5371G>A, el fragmento amplificado fue de 111 pares de bases con la utilización del iniciador sentido 5’ CCCTCCGTCCCATAGCAAA 3’ y del iniciador antisentido 5’ GTGGAGGACTTCACCGAGTT 3’. Los fragmentos amplificados fueron digeridos con las enzimas HpaII y HaeIII, respectivamente, de acuerdo con el programa NEBcutter V2.0 (New England Biolabs).13 En ambos casos, las enzimas reconocieron el alelo silvestre y la digestión se realizó a 37 °C en un plazo de 16 a 24 horas, con una inactivación a 80 °C por 20 minutos. Los fragmentos de digestión fueron observados mediante geles de poliacrilamida y AgNO3 (Figura 1).
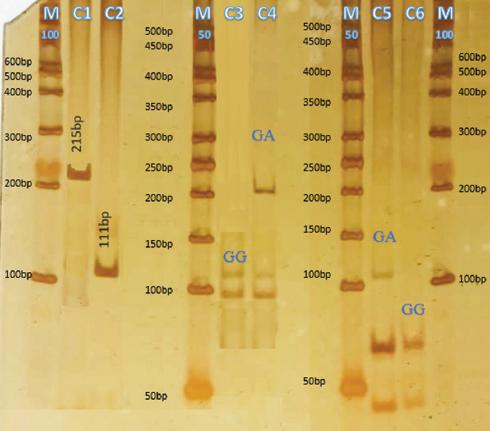
Figura 1 Electroforesis de los productos de PCR y digestión enzimática en gel de poliacrilamida a 6 % teñido con AgNO3. M100 se refiere al uso de un marcador de pares de bases (pb) y cada fragmento visible equivale a 100 pb; se aprecian fragmentos desde 100 pb hasta 600 pb. M50 se refiere al uso de un marcador de pb y cada fragmento visible equivale a 50 pb; se aprecian fragmentos desde 50 bp hasta 500 bp. C1 muestra el fragmento amplificado de c.3915G>A. C2 muestra el fragmento amplificado de c.5371G>A. C3 y C4 indican los fragmentos digeridos de c.3915G>A con el genotipo encontrado. C5 y C6 indican los fragmentos digeridos de c.5371G>A con el genotipo encontrado. Para evitar un error en la metodología ocasionado por una posible digestión parcial, se utilizó el fago lambda al no encontrar el genotipo AA, con lo cual se pudo verificar que efectivamente la digestión fue correcta y el genotipo no estaba presente en la población analizada.
Análisis estadístico e in silico
El análisis estadístico se realizó con el programa SPSS versión 25 (IBM Corp., Armonk, New York, Estados Unidos). Se estableció la distribución de las frecuencias alélicas y genotípicas de las dos variantes, que se compararon entre grupos mediante prueba de chi cuadrada. Se realizaron cuatro modelos de herencia (codominante, dominante, sobredominante y recesivo) y se calcularon las razones de momios (RM) y los intervalos de confianza de 95 % (IC 95 %) correspondientes para medir las asociaciones. El análisis de haplotipos se efectuó con el programa Arlequin ver 3.5.2.2 (Genetics Software, Berna, Suiza).14 El criterio de significación estadística fue p < 0.05. Para cada variante se realizó un análisis predictivo in silico con el programa Human Splicing Finder (Genomnis SAS, Marsella, Francia).15
Aprobación ética
El estudio fue aprobado por el comité de ética local del Centro Universitario del Sur (número 05-2010-1-853) de acuerdo con la Declaración de Helsinki y las guías nacionales de la Ley General de Salud de México en Materia de Investigación en Salud, título segundo, artículo 17, además de los artículos 13 a 21 (publicada el 22 de junio de 2017 en el Diario Oficial de la Federación).
Resultados
La muestra total estuvo integrada por 153 varones (59 %) y 107 mujeres (41 %), la edad promedio fue de 59.4 ± 7.2 años en el grupo con CCR y de 40.6 ± 10.4 en el grupo de control (Tabla 1); ambos grupos fueron pareados por sexo, pero no por edad. Las frecuencias alélicas y genotípicas de las variantes c.3915G>A y c.5371G>A, así como los modelos de herencia se describen en la Tabla 2. En los modelos codominante y sobredominante de la variante c.3915G>A se observó que el genotipo GA muestra una disminución del riesgo de CCR en comparación con el grupo de control (RM = 0.29, IC 95 % = 0.13-0.64, p = 0.002), al igual que el alelo A (RM = 0.31, IC 95 % = 0.15- 0.69, p = 0.004). Es importante resaltar que el genotipo AA de esta variante no se observó en ningún grupo. Respecto a la variante c.5371G>A no se encontraron diferencias significativas entre ambos grupos.
Tabla 1 Características demográficas de los pacientes con cáncer colorrectal y de los sujetos del grupo de control
| Variable | Con cáncer colorrectal (n = 126) | Grupo de control* (n = 134) | ||
|---|---|---|---|---|
| Media ± DE | Media ± DE | |||
| Edad en años | 58.9 ± 9.2 | 40.6 ± 10.1 | ||
| n | % | n | % | |
| Mujeres | 52 | 41.3 | 55 | 41.0 |
| Hombres | 74 | 58.7 | 79 | 59.0 |
*Sujetos sanos. DE: desviación estándar.
Tabla 2 Frecuencias alélicas y genotípicas de las variantes c 3915G>A y c. 5371G>A del gen TSC2 en pacientes con cáncer colorrectal y en los sujetos del grupo de control, conforme a cuatro modelos de herencia
| Modelo de herencia | Alelos | CCR (n = 126) | Grupo de control* (n = 132) | RM | IC 95 % | p | |||
|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | ||||||
| c. 3915G > A | Codominante | GG | 117 | 92.9 | 106 | 79.1 | 1 | Referencia | - |
| GA | 9 | 7.1 | 28 | 20.9 | 0.29 | 0.13-0.64 | 0.002 | ||
| AA | 0 | 0 | 0 | 0 | 0.9 | 0.02-46.08 | 0.960 | ||
| Dominante | GG | 117 | 92.9 | 106 | 79.1 | 1 | Referencia | - | |
| GA + AA | 9 | 7.1 | 28 | 20.9 | 0.29 | 0.13-0.64 | 0.002 | ||
| Recesivo | GG + GA | 126 | 100 | 134 | 100 | 1 | Referencia | - | |
| AA | 0 | 0 | 0 | 0 | 1.06 | 0.02-53.99 | 0.976 | ||
| Sobredominante | GG + AA | 117 | 92.9 | 106 | 79.1 | 1 | Referencia | - | |
| GA | 9 | 7.1 | 28 | 20.9 | 0.29 | 0.13-0.64 | 0.002 | ||
| G | 243 | 96.4 | 240 | 89.5 | 1 | Referencia | - | ||
| A | 9 | 3.6 | 28 | 10.5 | 0.31 | 0.15-0.69 | 0.004 | ||
| Modelo de herencia | Alelos | CCR (n = 122) | Grupo de control (n = 122) | RM | IC 95 % | p | |||
| n | % | n | % | ||||||
| c. 5371G > A | Codominante | GG | 117 | 95.9 | 117 | 95.9 | 1 | Referencia | - |
| GA | 5 | 4.1 | 5 | 4.1 | 1.0 | 0.28-3.55 | 1.000 | ||
| AA | 0 | 0 | 0 | 0 | 1.0 | 0.02-50.82 | 1.000 | ||
| Dominante | GG | 117 | 95.9 | 117 | 95.9 | 1 | Referencia | - | |
| GA + AA | 5 | 4.1 | 5 | 4.1 | 1.0 | 0.28-3.55 | 1.000 | ||
| Recesivo | GG + GA | 126 | 100 | 134 | 100 | 1 | Referencia | - | |
| AA | 0 | 0 | 0 | 0 | 1.0 | 0.02-53.99 | 0.976 | ||
| Sobredominante | GG + AA | 117 | 95.9 | 117 | 95.9 | 1 | Referencia | - | |
| GA | 5 | 4.1 | 5 | 4.1 | 1.0 | 0.28-3.55 | 1.000 | ||
| G | 239 | 97.9 | 239 | 97.9 | 1 | Referencia | - | ||
| A | 5 | 2.1 | 5 | 2.1 | 1.0 | 0.29-3.49 | 1.000 | ||
*Sujetos sanos.
Las variables fueron comparadas con la prueba de chi cuadrada. Se tomó en cuenta como valor de referencia el genotipo y el alelo más frecuente de cada variante. Los valores en negritas indican los datos estadísticamente significativos. Se consideró una p < 0.05.
CCR: cáncer colorrectal; IC 95 %: intervalo de confianza de 95 %; RM: razón de momios.
Las distribuciones genotípicas de las dos variantes examinadas en este estudio estaban en equilibrio de Hardy-Weinberg (p > 0.05, datos no mostrados).
De acuerdo con el análisis de haplotipos (Tabla 3), el haplotipo A/G, en el orden c.3915G>A/c.5371G>A, mostró un posible efecto protector contra CCR (RM = 0.28, IC 95% = 0.12-0.68, p = 0.005).
Tabla 3 Análisis de haplotipos de las variantes c. 3915G>A y c. 5371G>A del gen TSC2 en muestras de pacientes con cáncer colorrectal y grupo de control
| Haplotipo c. 3915G>A/ c. 5371G>A | CCR (n = 100) | Grupo de control* (n = 107) | RM | IC 95 % | p |
|---|---|---|---|---|---|
| G/G | 0.944 | 0.867 | 1 | Referencia | - |
| G/A | 0.016 | 0.016 | 0.98 | 0.19-4.91 | 0.979 |
| A/G | 0.036 | 0.114 | 0.28 | 0.12-0.68 | 0.005** |
| A/A | 0.004 | 0.003 | 0.98 | 0.06-15.77 | 0.988 |
Las variables fueron comparadas con la prueba de chi cuadrada.
*Sujetos sanos.
**Representa una diferencia significativa entre los pacientes con CCR y los individuos del grupo de control (p < 0.05).
CCR: cáncer colorrectal; IC 95%: intervalo de confianza de 95%; RM: razón de momios.
Finalmente, el análisis in silico mostró que la variante c.3915G>A crea un sitio silenciador de empalme exónico, lo que puede generar una modificación en el empalme alternativo, con el resultado de una proteína trunca o fácilmente degradable, lo cual probablemente altera su funcionalidad. Por otro lado, la presencia del alelo A de c.5371G>A activa un sitio aceptor críptico exónico y un sitio donante críptico exónico, que también pueden ocasionar modificaciones del corte y el empalme (Tabla 4).
Tabla 4 Análisis in silico de variantes del gen TSC2 humano
| Variante c 3915G>A | Variante c 5371G>A | ||
|---|---|---|---|
| Posición | 16:2083816-2083729: + | 16:2088547-2088560: + | 16:2088551-2088559: + |
| Secuencia | AGGGAAGTCCGGGC | CTATGAGGTGGGCC | GAGGTGGGC |
| Localización y cambio de nucleótido | 16 2083726 g/a | 16 2088557 g/a | 16 2088557 g/a |
| Señal | Nuevo aceptor del sitio de empalme. | Nuevo aceptor del sitio de empalme. | Nuevo donador del sitio de empalme. |
| Variación | 44.66 > 72.53 (62.4 %) | 38.78 > 66.65 (71.87 %) | 6.32 > 8.7 (37.66 %) |
| Puntuación | 72 | 66 | 8 |
| Fuerza | Débil | Débil | Media |
| Interpretación | — Activación de un sitio aceptor críptico exónico.
— Alteración potencial del corte y empalme. |
— Activación de un sitio aceptor críptico exónico.
— Alteración potencial del corte y empalme |
— Activación de un sitio donador críptico exónico.
—Alteración potencial del corte y empalme. |
Discusión
Las variantes en el heterodímero TSC1/TSC2 están asociadas a esclerosis tuberosa; estudios recientes han sugerido que TSC2 es un regulador celular negativo importante de la vía de señalización mTOR, que participa en procesos asociados al desarrollo de tumores.16
En el presente estudio, las frecuencias genotípicas de la variante c.3915G>A en el grupo de control son diferentes a las reportadas en la base de datos HapMap (mapa de haplotipos) y en el Proyecto 1000 Genomas, en mexicanos de Los Ángeles (G = 0.959 y A = 0.041), caucásico-estadounidenses (G = 0.94 y A = 0.006) y poblaciones europeas (G = 0.998 y A = 0.002). Sin embargo, fueron más cercanas a las encontradas en población luhya de Kenia (G = 0.867 y A = 0.133). Estas cifras pueden explicarse debido al alto mestizaje de la población indígena mexicana con los aproximadamente 250 000 africanos traídos en los tiempos de la Colonia; Kenia fue uno de los lugares de procedencia. Actualmente existen dos millones de afrodescendientes en México y es de estimar que un gran porcentaje de individuos no reconocidos como afrodescendientes posean características genotípicas africanas.17,18 Respecto a c.5371G>A, la frecuencia genotípica del alelo G fue de 0.979 y la del alelo A de 0021 en el grupo de control; el alelo A estuvo ausente en los pacientes con cáncer colorrectal.19-22
Como lo muestra el análisis in silico, la activación de un sitio aceptor críptico de la variante c.3915G>A parece modificar tanto la secuencia intrónica como el proceso de empalme, lo que puede traducirse en diferentes modificaciones del empalme, como los silenciadores de empalme exónico, los cuales regulan el empalme alternativo, con lo que se producen dos o más isoproteínas con funciones distintas.23 De acuerdo con nuestros resultados, sugerimos que la presencia del alelo A de la variante c.3915G>A favorece el empalme del ARNm que se traduce en una ventaja proteica. Los estudios funcionales podrían ayudar a determinar si esta variante tiene un efecto sobre la síntesis o expresión de la proteína.22,24-26 Por otro lado, el alelo A de la variante c.3915G>A se observó únicamente en heterocigosis y con una frecuencia muy baja en ambos grupos, lo que sugiere un posible efecto protector contra el desarrollo de CCR (p = 0.004). Sin embargo, debido a que no existen reportes previos al respecto, será necesario confirmar estos datos, principalmente en tejido tumoral, así como su posible relación con variantes en el gen APC.
La variante c.5371G>A ha sido previamente identificada en forma heterocigota en muestras de tejido de melanoma maligno.26 Hasta el momento, nuestro estudio es el primero en reportar las frecuencias genotípicas de la variante en población sana, así como en pacientes con CCR; observamos que en la mayoría de los sujetos del grupo de control se encontró de manera heterocigota y que estuvo ausente el genotipo homocigoto en el alelo A, tanto en el grupo de control como en los pacientes con CCR.
La variante sin sentido c.5371G>A genera el cambio de la glicina a serina y se encuentra justo al final de TSC2, muy cerca del gen PDK1. Los resultados del análisis in silico mostraron que el alelo A activó un sitio donante críptico exónico con un efecto potencial sobre el empalme. Debido a que el genotipo GA se observó en solo cinco pacientes (4.1 %) con CCR y en cinco individuos del grupo de control (4.1 %), no fue posible estimar su efecto sobre el desarrollo de CCR, lo cual resultó ser una limitación del estudio. Posiblemente, el seguimiento de la enfermedad en los individuos con presencia del genotipo GA pudiera proporcionar más información respecto al impacto de la variante en la evolución del CCR.26 Finalmente, se observó que el haplotipo A/G de las variantes c.3915G>A y c.5371G>A, respectivamente, tiene un posible efecto protector en la población con CCR; por lo que podemos concluir que el haplotipo con los alelos ancestrales no brinda protección ni reduce el riesgo de CCR.
Conclusiones
El presente estudio muestra que el alelo A de la variante c.3915G>A de TSC2 tiene un posible efecto protector contra CCR, al igual que el haplotipo A/G de las variantes c.3915G>A y c.5371G>A. Sin embargo, debido al tamaño muestral, será necesario realizar más estudios en una población mayor de las mismas regiones geográficas para obtener resultados con una fuerza estadística más robusta, ya que la comparación de sujetos de poblaciones distintas (en los casos o en los sujetos de control) podría derivar en conclusiones incorrectas. Asimismo, el análisis in silico señala que la presencia de las variantes puede implicar una modificación del proceso de corte y empalme que cambie la expresión de la proteína en pacientes con CCR.

