











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkAntecedentes
El angioedema hereditario (AEH) es una enfermedad rara de transmisión autosómica dominante que se manifiesta con episodios de angioedema espontáneo impredecible, edema cutáneo, de submucosas y de la vía aérea, que puede representar un riesgo para la vida del paciente (Cuadro 1). Se produce por deficiencia o disfunción del inhibidor C1 (C1-INH)1 y se presenta en alrededor de 1 entre 30 000 a 80 000 individuos de la población general. La enfermedad muestra un patrón autosómico dominante; 50 % en hombres y 50 % en mujeres. Los hijos de padres con AEH son afectados en la misma proporción.2
Cuadro 1 Criterios diagnósticos del AEH6
|
Criterios clínicos • Angioedema subcutáneo no inflamatorio, autolimitado, sin urticaria, recurrente y de duración > 12 horas. • Dolor abdominal autolimitado sin etiología orgánica, recurrente y de duración > 6 horas. • Edema laríngeo recurrente. |
|
Criterios
analíticos • Niveles de inhibidor C1 < 50 % de los niveles normales en 2 determinaciones después del primer año de vida. • Función del inhibidor C1 < 50 % de lo normal en 2 determinaciones después del primer año de vida. • Mutación del gen del inhibidor C1 (este criterio solo puede ser usado para el diagnóstico en pacientes < 1 año). • Niveles bajos de C4 durante las crisis, habitualmente normales en periodos intercrisis. |
*El diagnóstico requiere un criterio clínico y un criterio de laboratorio.
Existen tres tipos básicos de AEH:3
El tipo 1 es el más frecuente (85 %) y se debe a déficit del inhibidor C1 (proteína inhibidora de la proteasa serina); los niveles bajos de C1-INH y C4 sugieren AEH por déficit C1-INH en todas las edades.
El tipo 2 se debe a déficit funcional del inhibidor C1, con niveles normales o incluso elevados de la misma proteína.
El tipo III tiene una presentación clínica similar a los tipos I y II, pero no se debe a deficiencia o alteración funcional de C1-INH.
El déficit cualitativo o cuantitativo de C1-INH ocasiona activación anormal de la vía clásica del sistema de complemento, con consumo de los factores C4 y C2. Durante dicha activación se liberan mediadores vasoactivos que aumentan la permeabilidad capilar y desencadenan el angioedema.4
La sospecha clínica de C1-INH-AEH por síntomas a cualquier edad es una indicación para realizar evaluación en este sentido, sin importar la historia familiar. Como sucede con numerosos desórdenes autosómicos dominantes, puede tratarse de una mutación de novo que puede pasar a futuros descendientes.5
Tratamiento
El manejo del AEH incluye tratamiento agudo y profiláctico para prevenir o minimizar el número y severidad de ataques. Los objetivos del tratamiento agudo son minimizar la morbilidad y prevenir la mortalidad. El objetivo del tratamiento profiláctico es reducir la probabilidad de inflamación y la severidad de las crisis (profilaxis a largo plazo).7
El tratamiento farmacológico comprende concentrados plasmáticos derivados del inhibidor de C1, entre ellos Berinert® (CSL Behring, Alemania), aprobado para el tratamiento de ataques agudos de HEA en todas las edades, con dosis de 20 U/kg de peso corporal, vía intravenosa. Cinryze® (Shire Pharmaceutical) - derivado del plasma humano, con dosis de 1000 U de aplicación intravenosa (dos frascos), independientemente del peso corporal- está aprobado para el tratamiento de ataques agudos de AEH, así como para profilaxis a corto y largo plazos. Icatibant es un antagonista selectivo del receptor b2 de bradicinina. Ecallantide es una proteína recombinante de 60 aminoácidos que inhibe reversiblemente la actividad de la calicreína.
Los antifibrinolíticos (ácido ε-aminocaproico o ácido tranexámico) se sintetizan químicamente y ejercen su acción en AEH inhibiendo la conversión de plasminógeno a plasmina. En muchas partes del mundo, los derivados de andrógenos (metiltestosterona, danazol, estanozolol y oxandrolona) se utilizan para la profilaxis a corto y largo plazos. Los niveles plasmáticos de C1-INH se elevan con la administración de andrógenos atenuados, que proporcionan una explicación parcial para su efecto.8 Los agentes mencionados son eficaces y de bajo costo, pero pueden causar una variedad de eventos adversos; la dosis de danazol no influye en los parámetros hematológicos.9
El plasma fresco congelado contiene inhibidor de C1 y varios estudios no controlados han reportado beneficio con su uso en las crisis agudas ocasionadas por AEH, sin embargo, su uso es controvertido porque contiene proteínas del sistema de complemento, que puede proporcionar el sustrato para la generación adicional de bradicinina, exacerbando los síntomas.10
Caso clínico
Mujer de 16 años sin antecedentes personales o familiares de trastorno inmunológico o alérgico. Presentó episodios recurrentes (aproximadamente en 5 ocasiones) de edema facial, manos y miembros inferiores (Figura 1) de un año de evolución, de inicio súbito, indoloros, no pruriginosos, de 24 a 48 horas de duración. Fue tratada con diversos esquemas de antihistamínicos y corticosteroides, sin mejoría de los síntomas. En numerosas ocasiones, los episodios se acompañaron de edema laríngeo y asfixia con cianosis, que para su manejo han requerido la hospitalización de la paciente y han puesto en peligro su vida.

En la visita médica ambulatoria se realizaron estudios hematoinmunológicos: leucocitos 9400/mm3, neutrófilos 55 %, linfocitos 33 %, eosinófilos 2.1 %. Se investigó factor alergénico desencadenante por examen de heces, negativo para parásitos, al igual que los exámenes de orina; la inmunoglobulina E fue de 139 mg/dL. Los anticuerpos antinucleares (ANA) y anticuerpos antimitocontriales (ANCA) resultaron negativos. Los niveles de complemento fueron normales: C3, 153 µg/µL; C4, 12 µg/µL. En la segunda visita, la paciente mostraba remisión leve de los síntomas, por lo que se le aplicaron dos unidades de plasma fresco congelado; inmediatamente presentó exacerbación severa, angioedema generalizado y disfonía (Figura 2). A falta de respuesta con difenhidramina intravenosa, se le aplicaron 0.3 mL de adrenalina intramuscular, sin mejoría de los síntomas. El edema persistió durante 72 horas. La paciente permaneció en observación, sin que requiriera intubación endotraqueal (Figura 2).
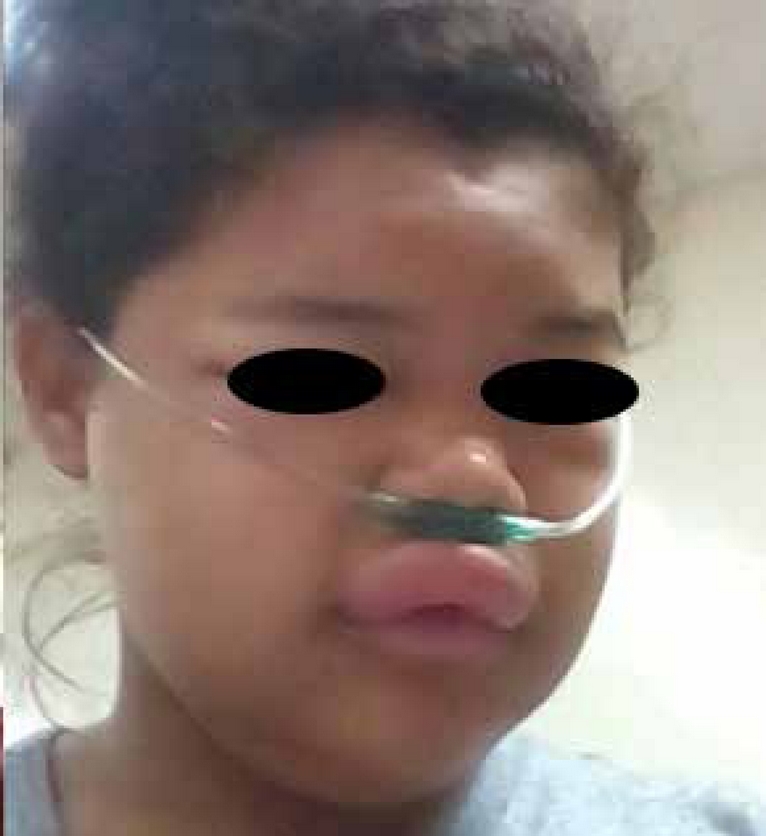
Se solicitó cuantificación de los niveles del inhibidor C1 esterasa en el extranjero, ya que este estudio no está disponible en el país. Los niveles fueron bajos: 7.1 µg/µL (24-39 µg/µL), con lo que se confirmó el diagnóstico de angioedema por déficit del inhibidor C1. Se comenzó tratamiento profiláctico con 600 mg/día de danazol durante 7 días, hasta disminuir la dosis de mantenimiento a 200 mg/día. Al momento de este reporte se había reducido el número de exacerbaciones: un episodio leve de angioedema por año.
Discusión
El diagnóstico de AEH es similar en adultos y niños. Los eventos clínicos en pacientes con C1-INH-AEH se caracterizan por episodios recurrentes subcutáneos o submucosos edematosos sin erupciones ni prurito, cuya duración sin tratamiento puede prolongarse de 1 a 5 días antes de la resolución espontánea.11 El estudio que se realiza por sospecha de AEH es la cuantificación de los niveles séricos de los inhibidores C1 y C4.
El retraso diagnóstico es común en los pacientes con AEH; a menudo se descarta por historia familiar negativa, sin embargo, aproximadamente 25 % de los pacientes con AEH presenta mutación nueva del inhibidor C1, no manifestada en su herencia.12 La paciente de este caso no tenía antecedentes inmunológicos o alérgicos que orientaran el diagnóstico, por lo tanto, es muy probable que la mutación de novo del inhibidor C1 se manifestara efectivamente.
Existen múltiples fármacos para el tratamiento tales como Berinert, icatibant, ecallantide y Cynrise, que se administran durante las manifestaciones agudas y como profilaxis a largo plazo. En la paciente descrita no pudieron ser aplicados por dificultades sanitarias de la región; además, estos fármacos son inaccesibles económicamente, sin contar que no existen casos registrados de AEH en la región. Para el tratamiento profiláctico a largo plazo del AEH se utilizan anabólicos con baja actividad androgénica, como el danazol, agentes antifibrinolíticos como los ácidos épsilon aminocaproico y tranexámico o el concentrado plasmático del inhibidor C1 derivado del plasma, que disminuyen la gravedad y frecuencia de los ataques agudos.13
Cuando los medicamentos derivados de concentrado plasmático del inhibidor C1 no están disponibles o accesibles, en una crisis aguda está indicado el uso de plasma fresco congelado, la dosis es de 10 mL/kg, pero puede administrarse a demanda (nivel de seguridad, evidencia III). La respuesta de nuestra paciente coincidió con la referida en revisiones donde se describe riesgo de exacerbación de los síntomas.5 En ella se indicaron las características propias de la enfermedad con variante de mutación nueva sin antecedentes. Debido a restricciones sanitarias no se dispuso de derivados plasmáticos del inhibidor C1, por lo que se aplicó plasma fresco congelado, que agudizó la reacción. El uso profiláctico de andrógenos, como el danazol (opción al alcance de los países en desarrollo), ha disminuido considerablemente el número y severidad de las crisis en el último año, con notable mejoría de la calidad de vida de la paciente.

