











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkLos hidrocarburos aromáticos policíclicos (HAP) son un grupo de más de 100 sustancias químicas diferentes, que se forman durante la combustión incompleta de la materia orgánica y son liberados al ambiente en grandes cantidades1,2. Debido a su persistencia y toxicidad la Agencia de Protección del Medio Ambiente de los Estados Unidos (EPA) ha incluido a 16 de estos HAP (Cuadro 1) en su lista de contaminantes orgánicos persistentes3.
Cuadro 1 Hidrocarburos aromáticos policíclicos (HAP) de acuerdo a su peso molecular, según la Agencia de Protección Ambiental de los Estados Unidos (EPA)
| HAP | Abreviación | Peso molecular (g/mol) |
|---|---|---|
| Bajo peso molecular (BPM) | ||
| Naftaleno | NAP | 128 |
| Acenafteno | ANA | 154 |
| Acenaftileno | ANY | 152 |
| Fluoreno | FLU | 166 |
| Fenantreno | PHE | 178 |
| Antraceno | ANT | 178 |
| Alto peso molecular (APM) | ||
| Fluoranteno | FLT | 202 |
| Pireno | PYR | 202 |
| Benzo(a)antraceno | BaA | 228 |
| Criseno | CHR | 228 |
| Benzo(b)fluoranteno | BbF | 252 |
| Benzo(k)fluoranteno | BkF | 252 |
| Benzo(a)pireno | BaP | 252 |
| Benzo(g,h,i)perileno | BPE | 276 |
| Indeno (1,2,3-cd)pireno | IPY | 276 |
| Dibenzo(a,h)-Antraceno | DBA | 278 |
EPA, 19983.
La presencia de estos compuestos en el aire en forma de material particulado fue informada a nivel global4, y también sus depósitos y acumulación en suelos y pastos5,6. Cuando los pastos son consumidos por vacas en estado de lactación, se ha demostrado su presencia en leche y derivados lácteos7-11. La contaminación de leche con HAP, depende de factores ambientales, como son: la fuente de exposición, la etapa de lactancia, el estado de salud del animal y del sistema de crianza12,13.
El consumo de leche con HAP representa un riesgo para la salud humana por lo que la Unión Europea (UE), ha establecido un máximo de residuos para la presencia de benzo(a)pireno (BaP) y la sumatoria de cuatro de ellos: BaP, benzo(a)antraceno (BaA), benzo(b)fluoranteno (BbF) y criseno (CHR) en diferentes alimentos que oscila entre 1 a 35 µg kg de grasa14.
No existe un método oficial para la determinación de HAP en leche, existiendo dos tendencias en la identificación y cuantificación de los mismos: 1) por cromatografía gaseosa empleando detector de ionización de flama y espectrometría de masas15,16, 2) por cromatografía de líquidos de alta resolución con detector de fluorescencia7,8,17. Para obtener mejores resultados en la preparación de las muestras se han empleado diferentes procedimientos que incluyen saponificación, extracción líquido-líquido (LLE) y limpieza por cromatografía en columna o más recientemente, extracción en fase sólida (SPE)18,19,20.
La determinación directa de HAP en leche mediante saponificación y extracción posterior, o la extracción de la grasa seguida de purificación muestran perfiles diferentes de los HAP en leche, con un predominio de fenantreno (PHE), antraceno (ANT), fluoreno (FLU), pireno (PYR), BaA y CHR. Por lo anterior el objetivo del presente trabajo fue determinar la presencia de HAP en marcas de leche evaluando tres procedimientos de extracción.
Se seleccionaron de manera aleatoria cuatro marcas comerciales de leche entera (tres ultrapasteurizadas (UHT) y 1 pasteurizada (HTST); cada marca tuvo tres muestreos para una n= 12; las muestras se colectaron durante el periodo marzo-junio del 2016 en supermercados ubicados en la Ciudad de México en la delegación de Coyoacán. Las muestras de leche UHT y pasteurizada se almacenaron en el laboratorio de análisis instrumental de la Universidad Autónoma Metropolitana-Xochimilco, las primeras en un lugar seco y fresco, la pasteurizada en refrigeración a 5 °C hasta su análisis, el cual no superó los cinco días después de su compra. Las muestras se homogeneizaron a baño maría (40 °C) por 30 min y agitadas manualmente cada 5 min antes de iniciar el proceso de extracción.
Variante A: Saponificación de la leche. De acuerdo al método de Girelli et al17 modificado: a 4 ml de leche (4 g) se le agregaron 8 ml de una solución de hidróxido de sodio en etanol (0.4 M); la mezcla se homogeneizó por un minuto en vortex y se colocó en un baño térmico a 40 0C hasta casi sequedad (1 ml). Se llevó a sequedad bajo una corriente de nitrógeno. Finalmente se reconstituyó en 1,000 µl de isooctano y se almacenó a -20 0C hasta su análisis.
Variante B: Extracción con solución detergente. En un matraz volumétrico de 500 ml se adicionaron 250 ml de muestra más 250 ml de una solución detergente (50 g de hexametafosfato de sodio en 24 ml de Tritón X -100 disueltos en un litro de agua). El matraz se agitó vigorosamente colocándose en un baño de agua a 90 0C invirtiéndose cada 15 min hasta lograr una separación en el cuello del matraz de la materia grasa. La grasa extraída se filtró a 50 0C a través de un papel filtro Whatman número 4, en presencia de sulfato de sodio anhidro y se conservó en tubos de vidrio a -20 oC hasta su análisis21.
Variante C: Extracción líquida-líquida (AOAC 989.05). En un embudo de separación se adicionaron 150 ml de muestra y 0.5 g de ácido etilendiaminotetraacético (EDTA), agitándose por un minuto y se reposó por 2 min. Se adicionaron 50 ml de metanol, volviéndose a agitar por un minuto. Se repitió la operación, adicionándose 50 ml de éter dietílico y 50 ml de éter de petróleo. Se dejó en reposo hasta la separación de la fase orgánica (sobrenadante), se drenó la capa inferior y el sobrenadante, pasándose por papel filtro Whatman número 1, adicionando 5 g de sulfato de sodio anhidro. La capa orgánica se rotovaporó a 40 0C y se traspasó a un frasco de 5 ml y fue guardado a -20 0C hasta su análisis.
La muestra de grasa (saponificada) se depositó lentamente sobre una columna que contenía 6 g de sílica gel (en la parte inferior) y 1 g de sulfato de sodio anhidro. Se adicionaron 20 ml de hexano, la fase orgánica se marcó como F1. Se cambió el matraz y se adicionaron 30 ml de hexano-diclorometano 9:1 v/v, se dejó correr lentamente y al llegar al ras del sulfato de sodio se adicionaron 20 ml de hexano-diclorometano 1:1 v/v. Toda la fase orgánica se colectó en un solo matraz y se marcó como F2 (hidrocarburos aromáticos policíclicos recobrados). El extracto F2 se rotovaporó a 40 0C hasta casi sequedad (1 ml). La muestra se traspasó a un vial color ámbar, se llevó a sequedad bajo una corriente de nitrógeno, se reconstituyó en 250 µl de isooctano y se almacenó a -20 0C hasta su análisis19.
Se utilizó un cromatógrafo de gases digital de alta resolución modelo Shimadzu GC 2010 con automuestreador, un inyector PTV a 250 0C en modo Splitless con un sampling time (1 min), purga Flow 5.0 ml min-1, Septum purga 5 ml min-1. Como gas transportador se usó nitrógeno a un flujo de 9.8 ml min-1. Se trabajó con una columna HP5-MS (30 m x 0.025 mm DI. X 0.25 µm de grosor). El programa de temperatura del horno se estableció de la siguiente manera: temperatura inicial de 40 0C por 3 min, luego un incremento de 2 oC/ min hasta los 50 0C, de los 50 0C a los 160 0C con un aumento de 3 oC/ min, de los 160 0C a los 210 0C con un aumento de 5 0C/ min, de 210 a 255 0C con un incremento de 7 oC/ min, de 255 a 265 0C, con un aumento de 4 oC/ min. Por último, de 265 a 300 0C un incremento de 5 oC/ min, manteniéndose por 5 min. Para el análisis de los cromatogramas se empleó el Sotware GG solution.
Se empleó un cromatógrafo Agilent GC 5890, se aplicó 1 µl de extracto de la muestra mediante inyección (en columna). Se utilizó columna capilar Rtx-5Sil MS (30 m, x 0.25 mm DI, 0.25 µm de grosor) de Restek (Bellafonte, PA, EE. UU.) y una precolumna Siltek de 2 m (0.53 mm DI.) del mismo proveedor. Se usó helio como gas transportador a un flujo constante de 1 ml/min. La temperatura del inyector se estableció en 3 oC superior a la temperatura del horno en todo momento. La temperatura del horno se programó de la siguiente manera: 1 min a 100 oC, de 100 °C a 300 oC a 5 oC /min y 15 min a 300 oC. La detección de los analitos se realizó con un equipo Agilent MS 5972 en el modo de impacto de electrones con una energía de ionización de 70 eV y monitorización de ión único22 (Figura 1).
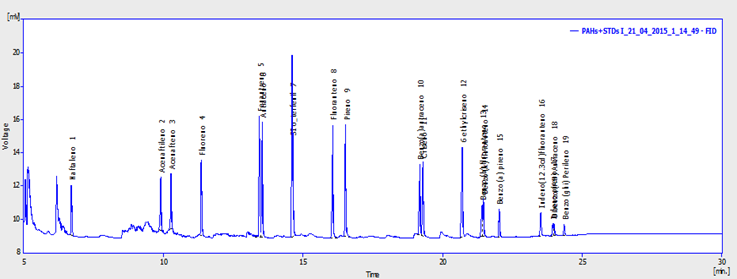
Estándar interno: ortoterphenyl (pico 7); 6 ethyl chrysene (pico 12); indeno[1,2,3-cd]fluoranthene (pico 16).
Figura 1 Cromatograma de espectrometría de masas, identificando un pico de la muestra para saber si corresponde a un compuesto nativo
Los reactivos químicos y solventes fueron de calidad reactivo y HPLC respectivamente, de la firma comercial J. T. Baker chemical, USA. Para la detección y cuantificación de los analitos, se utilizó una mezcla de patrones con 16 compuestos de HAP recomendados en el método EPA 610 (Chemicalservice, USA), a saber: naftaleno (NAP); acenaftaleno (ALC); acenaftileno (ACY); fluoreno (FLU); fenantreno (PHE); antraceno (ANT); fluoroantraceno (PMA); pireno (PYR); benzo(a)antraceno (BaA); criseno (CHR); benzo(b)fluoranteno (BbF); benzo(k)fluoranteno (BkF); benzo(a)pireno (BaP); dibenzo(ab)antraceno (DBA); benzo(ghi)perileno (BGP) e indeno (cd)pireno (IcdPy) (Cuadro 1).
La extracción por medio de la saponificación de la leche (variante A)17 identificó solamente HAP de BPM, mientras que la extracción con solución detergente (variante B)19 logró determinar HAP de BPM y APM. Para el método B estas últimas moléculas representaron el 33.33 % de la concentración total de HAP, en tanto que, compuestos de BPM representaron el 66.66 % (Cuadro 2).
Cuadro 2 Concentración (µg g-1) de hidrocarburos aromáticos policíclicos (HAP) en leche por dos métodos de extracción
| HAP | Método A | Método B |
|---|---|---|
| NAP | 0.066 | Nd |
| ALC | 0.200 | 0.372 |
| ACY | 0.066 | Nd |
| FLU | Nd | 0.915 |
| PHE | Nd | 7.153 |
| ANT | 5.385 | 14.924 |
| FLT | Nd | Nd |
| PYR | Nd | 3.773 |
| BaA | Nd | 0-.056 |
| CHR | Nd | 0.044 |
| BbF | Nd | 1.264 |
| BkF | Nd | 0.750 |
| BaP | Nd | 0-.114 |
| DBA | Nd | 4.061 |
| BGP | Nd | Nd |
| IcdPy | Nd | 1.641 |
| Suma de 16 HAPs | 5.717 | 35.067 |
| Suma de 4 HAPs | 0.0 | 1.478 |
| Suma de HAPs DE BPM | 5.717 (100 %) | 23.365 (66.6 %) |
| Suma de HAPs DE APM | 0.00 | 11.702 (33.4 %) |
Métodos= A: saponificación y extracción directa; método B: extracción con solución detergente.
Nd= no determinado; µg g-1: microgramo de HAP por gramo de grasa láctea.
En este estudio la extracción de HAP por saponificación de la leche (variante A), difiere de otros estudios que muestran un predominio de HAP de APM con mayores concentraciones de PHE y ANT, así como la relación HAP de BPM y total de HAP que se ha reportado entre 50 y 68 %(17, 23). La ausencia de compuestos de APM en el método A, puede deberse a que las muestras analizadas presentan bajas concentraciones, aspecto que ha sido mencionado por otros autores24, por otra parte el empleo de 4 ml de muestras de leche, no es suficiente para sobrepasar el límite de detección de los HAP en nuestras condiciones (detector de ionización de flama).
Fórmulas lácteas para infantes18,25, leche entera y UHT17 han reportado concentraciones bajas de HAP de APM. Por otra parte, compuestos de BPM (2 y 3 anillos) en particular NAP, ACE y ACY no se informan en diversos estudios9,17,23, ya que reportan porcentajes de recuperación menores a 50, posiblemente por su alta volatilidad17. Sin embargo, es probable que el tiempo y la temperatura en la que se realiza la saponificación, jueguen un papel importante en los recobrados del mismo, en este estudio, se aplicó una temperatura de 40 oC pudiendo detectar los compuestos de BPM, similar a cuando se realiza la saponificación a 60 oC 24, mientras la saponificación a 80 oC, solo detecta PHE y ANT23, lo que es evidencia, que la temperatura de saponificación es un punto crítico en la determinación de HAP9.
A diferencia del método anterior, cuando se emplea la grasa sin saponificar y se pasa a una columna para su purificación, los resultados obtenidos permiten identificar HAP de BPM y APM, en valores de 66.6 y 33.4 % respectivamente, con perfiles similares a cuando se estudiaron 31 muestras de leche procedente de Brasil y Argentina con valores promedios de 75.5 y 24.5 % para HAP de BPM y APM respectivamente7, mientras que en leche fresca procedente de granjas cercanas a una área industrial se obtuvieron valores de HAP de BPM que oscilaron entre 40 y 69 %19; las diferencias encontradas pueden estar relacionadas con la forma de extracción de la grasa láctea, que se fundamentan en una extracción con solventes orgánicos7 y otra con una solución detergente19.
La mayor parte de los estudios que emplean la saponificación directa en las muestras usan detectores acoplados a masas o detectores fluorescentes, lo que les permite cuantificar bajas concentraciones de HAP en las muestras17,18,24; sin embargo, para alcanzar la sensibilidad adecuada, cuando se usa cromatografía de gases con detección de ionización de flama, se requiere emplear una mayor cantidad de grasa láctea en la determinación, aspecto que es posible obtener por el uso de una solución detergente, aunque los HAP se pueden perder por efecto de la temperatura a la que se someten las muestras durante la extracción (90 oC).
En el Cuadro 3 se presentan los porcentajes de recuperación de HAP en las muestras analizadas de acuerdo a los métodos B y C y la confirmación por CG-MS. Los recobrados obtenidos muestran una variabilidad alta, en función de la clasificación de los HAP de BPM y APM, donde los recobrados mayores se obtienen para los de APM. La variabilidad de los recobrados entre BPM y APM está asociados con el tipo de extracción de la grasa y la temperatura en la rotovaporación. En las condiciones de este trabajo, el método más adecuado fue el método C, ya que alcanza recobrados que varían entre 45.3 a 95.1 %, similares a los encontrado en otro estudio donde se emplearon solventes orgánicos en la extracción de la grasa, reportando valores de recobrados entre 40 y 125 % sin especificar los compuestos26. Otro estudio, pero en leche humana el recobrado osciló entre 42 y 101 % justificando los mismos con su punto de ebullición encontrando una R2 de 0.77927. Un estudio en leche de polvo, pero empleando un baño de ultrasonido y posterior purificación en columna, los recobrados oscilaron entre 95 y 98 %15; lo mismo sucede cuando se emplea un sistema de microextracción de fase sólida donde los recobrados oscilaron entre 87.6 y 112 %28.
Cuadro 3 Porcentajes de recuperación de hidrocarburos aromáticos policíclicos (HAP) en leche empleando dos métodos de extracción (media ± error estándar)
| HAP | Método B | Método C |
|---|---|---|
| NAP | Nd | Nd |
| ALC | 15.2±7.3 | 45.3±19.0 |
| ACY | 10.8±9.1 | 46.5±14.7 |
| FLU | 23.8±4.8 | 72.3±20.9 |
| PHE | 28.3±10.7 | 67.6±22.6 |
| ANT | 30.4±15.3 | 61.6±16.9 |
| FLT | 48.0±10.9 | 77.5±24.8 |
| PYR | 44.7±1467 | 72.0±25.9 |
| BaA | 70.9±16.7 | 80.0±14.4 |
| CHR | 59.3±15.5 | 95.1±27.5 |
| BbF | 93.5±21.1 | 80.9±11.3 |
| BkF | 45.4±11.1 | 72.7±22.4 |
| BaP | 127.0±35.0 | 85.6±7.0 |
| DBA | 78.4±17.9 | 92.1±18.3 |
| BGP | 64.5±15.7 | 86.9±21.9 |
| IcdPy | 66.3±14.7 | 75.0±15.7 |
| Suma de HAPs de BPM | 15 ± 8 % | 58.7±12.3 |
| Suma de HAPs de APM | 58 ± 21 % | 81.8±8.0 |
Métodos= B: extracción con solución detergente, método C: extracción líquida-líquida.
Se demuestra que la forma de extracción de la grasa constituye un punto crítico en la determinación de HAP en muestras de leche, sin embargo, los recobrados encontrados en el método C, están acordes cuando se determinan contaminantes ambientales en matrices biológicas y aparecen en concentraciones inferiores a 1 µg kg-1 donde su intervalo puede ser desde -50 % a +20 %29, lo cual permite evaluar la presencia de HAP en muestras de leche con una exactitud adecuada.
El Cuadro 4 presenta la incidencia, sumatoria, valor máximo y mínimo de cada HAP en el total de muestras analizadas. Una de las marcas no presentó HAP, mientras que en el resto se detectaron al menos uno de los 16 compuestos, es decir, 75 % de las muestras fueron positivas a la presencia de HAP. Los compuestos con mayor incidencia en las muestras fueron PHE y ANT con 54.5 % y FLUO y DBA con 45.5 %, donde el ANT presentó la mayor concentración (341 µg g-1) seguido del PHE (20 µg g-1) y el DBA (12.3 µg g-1). Estos resultados coinciden con otros autores donde compuestos de BPM aparecen con mayor frecuencia y concentración(17, 23).
Cuadro 4 Presencia de hidrocarburos aromáticos policíclicos (HAP) en las muestras de leche analizadas (n=12)
| ACE | FLUO | PHE | ANT | PYR | BaA | CHR | BbF | BkF | BaP | IND | DBA | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Inc | 36.4 | 45.5 | 54.5 | 54.5 | 9.1 | 27.3 | 27.3 | 27.3 | 18.2 | 9.1 | 27.3 | 45.5 |
| Suma | 1.2 | 5.6 | 20.0 | 341.0 | 3.8 | 0.2 | 0.2 | 2.3 | 1.1 | 0.1 | 6.3 | 12.3 |
| Min | 0.2 | 0.3 | 0.4 | 0.0 | 3.8 | 0.0 | 0.0 | 0.2 | 0.3 | 0.1 | 0.7 | 0.1 |
| Max | 0.6 | 3.6 | 7.5 | 155.0 | 3.8 | 0.1 | 0.1 | 1.3 | 0.7 | 0.1 | 3.0 | 5.6 |
Inc= incidencia, Min= mínimo, Max= máximo.
Por otra parte, la marca D presentó la mayor mediana de la sumatoria de 4 HAP (Cuadro 5); esta concentración sobrepasa el valor establecido por la UE que es 1 µg kg-1(14 para fórmulas lactantes, lo que implica un riesgo para la salud humana. Esta misma marca incorpora grasa vegetal en su formulación.
Cuadro 5 Resultados de la mediana de la sumatoria de 16 y 4 hidrocarburos aromáticos policíclicos (HAP) en marcas de leche de mayor consumo en la Ciudad de México
| Marcas | Ʃ 16 HAPs µg kg-1 | Ʃ 4HAPs µg kg-1 |
|---|---|---|
| A | Nd | Nd |
| B | 47.56 | 0.23 |
| C | 93.95 | 1.14 |
| D | 51.49 | 4.04 |
Nd= No determinada.
Las variantes B y C durante la determinación de los ácidos en muestras de leche presentan variables en los porcentajes de HAP de bajo y alto peso molecular, donde la variante C mostró los mejores recuperados, mientras la variante B puede ser una alternativa cuando se emplea la CG-FID. El 75 % de las muestras de leche fueron positivas a la presencia de HAP, donde el 50 % superaron el valor umbral de la UE.

