











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Arterial hypertension has a global prevalence of 30% and is responsible for 10.4 million deaths annually1. In Mexico, the prevalence of hypertension in people over 20 years of age is 37.4%2. International guidelines consider optimal blood pressure (BP) for an adult as < 120 mmHg of systolic blood pressure (SBP) and < 80 mmHg of diastolic blood pressure (DBP). The American College of Cardiology defines hypertension as SBP ≥ 130 mmHg and/or DBP ≥ 80 mmHg3. High blood pressure can be secondary to numerous causes and if it does not have a specific etiology that is called primary (previously called "essential") hypertension. Primary hypertension is polygenic and is characterized by low-grade inflammation in the kidneys that may play a role in causing renal vasoconstriction that impairs sodium excretion and leads to elevated systemic BP. Diagnosis of primary hypertension rests on ruling out the known causes of secondary HTN, the latter which may represent up to 1/3 of the total incidence of hypertension4. The etiologic factors and mechanisms that play a role in primary hypertension are subjects of research interest because of their importance in global health.
Immune system and hypertension
Innate immunity, acquired immunity, and autoimmunity are associated with hypertension. Inflammation resulting from immune reactivity activates sodium transport systems in the kidney, impairs vasorelaxation in peripheral arterial system, and maintains an overactive sympathetic nervous system5. The activation of innate immunity in hypertension was initially suggested by the observation that toll-like receptor 4 (TLR4) is overexpressed and proinflammatory cytokines are produced by stimulating TLRs in the spontaneously hypertensive rat (SHR)6. Furthermore, TLR4-deficient mice are resistant to LNAME-induced hypertension7. This suggests that TLR, which have a role in driving innate immune responses through activation of the inflammasome, could be involved in animal models of hypertension.
Uric acid and HSP70 are, among others, danger associated molecular patterns (DAMPs) that bind TLR2 and TLR4 and activate the NLRP3 inflammasome, both appear to be involved in the pathogenesis of hypertension in the SHR rat5. In hypertensive patients, there is also evidence of activation of the inflammasome as noted by increased circulating levels of IL-1β and IL-18 that are correlated with the severity of hypertension8.
Acquired immunity is also demonstrated in experimental models of hypertension. Elegant studies have shown incorporation of putative antigens into antigen-presenting cells in situ and in antigen presenting dendritic cells in circulation9, costimulation10, and generation of memory T cells11. In addition, circulating antibodies to relevant autoantigens have been detected in patients with primary hypertension12. Endogenous antigens currently studied in humans are proteins modified by isoketals (γ-ketoaldehydes) resulting from lipid peroxidation9 and heat stress protein 70 (HSP70)13. Autoimmune reactivity directed against extracellular HSP70 has been shown in hypertensive patients and it has been suggested that may result from molecular mimicry between human HSPs and bacterial HSPs14.
The clinical relevance of immune reactivity in hypertension has been shown by the correction of severe resistant hypertension with immunosuppressive treatment reported in isolated patients14 that require confirmation in larger studies.
Hyperuricemia and hypertension
Hyperuricemia and its adverse effects have been the focus of considerable interest in the past decades. Several studies have shown that high uric acid levels are associated and may be a causal factor, in chronic kidney disease and hypertension15. Hyperuricemia results from an imbalance between the production and elimination of uric acid. Diet is an important factor that modulates uric acid concentrations. The intake of red meat, organ meats, shellfish, beer, hard wine, and sugar all have the effect of raising serum uric acid and thereby predisposing to gout. While this was once more common among the rich, the high production of sugar in the 20th century led to more affordability, and today hyperuricemia and gout are widely prevalent among those on a western diet, especially for those whose diet includes sugar sweetened items16,17. The mechanism by which sugar raises uric acid is due to it containing fructose, which generates uric acid from the breakdown of ATP that occurs during its metabolism18.
Uric acid is the end-product of the purine catabolism pathway and xanthine oxidase is the limiting enzyme for its synthesis. Uric acid is a weak acid (pKa = 5.7), so at blood pH (pH = 7.40), it is ionized, mostly being in the form of sodium urate. Uric acid is poorly soluble in aqueous solutions and its solubility limit is around 7 mg/dL in serum, although stable supersaturated solutions can be formed under certain conditions. Increasing concentrations of uric acid cause the generation of monosodium urate crystals that are deposited in different organs, particularly in the joints, the kidney, and blood vessels19.
The role of uric acid in hypertension was studied in rats using an experimental model of moderate hyperuricemia20. Rodents, as well as most mammals, have a low serum uric acid level (~ 1 mg/dL) due to the presence of the liver enzyme uricase, in contrast with higher primates that lack this enzyme and have higher levels of serum uric acid (> 3 mg/dL)21. In rats, moderate hyperuricemia may be induced by inhibition of uricase with low doses of potassium oxonate. Unlike other models of inhibition of this enzyme, which result in massive uricosuria with intrarenal crystal deposition and obstructive renal disease, low doses of oxonate induce moderate hyperuricemia without intrarenal crystal deposition20. In these rats, hypertension developed after 3 weeks of moderate hyperuricemia22 and blood pressure correlated positively with plasma uric acid concentrations23. Several mechanisms have been proposed to explain the increase in blood pressure mediated by hyperuricemia, including preglomerular vasoconstriction, increased oxidative stress, endothelial dysfunction, and, pertinent to the present discussion, pro-inflammatory reactivity22. As will be discussed later, uric acid crystals24 as well as soluble uric acid25 have been shown to activate the NLRP3 inflammasome.
Heat shock protein 70 in hypertension
HSPs are evolutionary preserved proteins that act as chaperones to other proteins preventing their aggregation and destruction. HSPs represent 5-10% of the total protein content of the cell and constitute a cytoprotective system of critical importance for biological homeostasis. Overproduction of HSPs results from activation of the promoter region of the HSP gene in response to physical, ischemic, metabolic, and toxic stress. After their chaperone function is accomplished, HSPs dissociate from their "client" proteins and their intracellular content is adjusted by transcriptional and post-transcriptional modulation. HSPs are intracellular proteins but they can be translocated to the extracellular space in circumstances of cell damage, or by extrusion mediated by exosomes and ectosomes, or in association with secretory lysosomes. HSPs are classified by their molecular weight and HSP70 is the best studied. HSP70 is an immunodominant molecule capable of stimulating or suppressing the inflammatory response14,23,24. The existence of autoimmune reactivity to HSP70 in humans may have pathogenic relevance since it has been demonstrated in the kidney and in arterial vessel walls in experimental models of hypertension (for review see26). In general, intracellular HSP70 suppresses the activation of the inflammasome and inflammasome-dependent inflammation while extracellular HSP70 activates the inflammasome27. HSP70 of pathogenic microorganisms act as a damage associated molecular patterns (DAMP) and trigger the innate immune response28 stimulating toll-like receptor TLR2. TLR4 and CD14 in monocytes, thereby promoting the activation of the inflammasome and the secretion of caspase, IL-1β, IL-18, IL-6, and TNFα29. Membrane-bound or extracellular autologous HSP70 can also be autoantigenic and play a role in the development of primary HTN. In the experimental models of salt-sensitive hypertension, HSP70 is overexpressed in the kidney30. Several studies have found high circulating levels of HSP70 and anti-HSP antibodies in hypertensive patients12,31,32. Circulating T lymphocytes of hypertensive patients react with an accelerated memory-like proliferative response when challenged with HSP7033.
The pro-hypertensive reactivity induced by extracellular HSP70 includes the activation of the inflammasome. HSP70 binds CD14 on the plasma membrane of human monocytes with high affinity and the resulting increased expression of IL-1β and is consistent with inflammasome activation34. Pons et al.19 studied the role of HSP70-induced autoimmunity in experimental hypertension (19). In these studies, autoimmune reactivity to HSP70 was manifested by a clonal T CD4+ response and renal inflammation that resulted in the development of salt sensitive HTN. In contrast, immune tolerance to HSP70 induced with a highly preserved sequence of the mycobacterial HSP70, generated T regulatory cells secreting IL-10, and prevented salt-sensitive hypertension13.
There are other potentially important functions of HSP70 related to the immune reactivity in hypertension. Intracellular HSPs, acting in their chaperone function, may preserve the integrity of other exogenous or endogenous antigenic proteins of pro-hypertensive relevance and facilitate their transport to the major histocompatibility complex in antigen presenting cells by canonical as well as cross-presentation pathways30.
HSP70 and hyperuricemia driving inflammasome activation in primary hypertension.
Inflammasomes are multimeric complexes that act as sensors of protein patterns and are mediators of the development of inflammation. To date, four subfamilies of inflammasomes have been described (NLRA, NLRB, NLRC, and NLRP). The NLRP subfamily has 14 members, including NLRP3 (Nod Like Receptor family Pyrin domain containing 3) inflammasome or cryopyrin, which is the most extensively studied. Inflammasome activation by danger associated molecular patterns (DAMPS) and pathogen associated molecular patters (PAMPS) represents the first line of non-specific, defense system, and the innate immunity24,35. Canonical activation of NLRP3 inflammasome is a two-step process that requires priming with NFkB that generates pro-caspase and pro-IL-1B and pro-IL-18 and the subsequent articulation of the inflammasome that may result from several pathways activated by variety of stimuli35. These pathways include binding of extra-cellular ATP to its receptor P2X7, leading to intracellular K+ efflux or, alternatively, direct NLRP3 activation by ROS or by Cathepsin B generated by lysosomal damage resulting from phagocytosis of crystalline structures36. Activation of the NLRP3 inflammasome complex results in the generation of active caspase, active cytokines, and pyroptosis, a type of cellular death caused by the formation of pores in the cellular membrane leading to cell edema, lysis, and extrusion of inflammatory cytokines. The pores are caused by caspase-induced gasdermin activation. The role of inflammasome activation in hypertension is underlined by the observations that pharmacological inhibition of NLRP3 and as well as individual deletion of its components prevents experimental hypertension37-39. In hypertensive patients, activation of the NLRP3 inflammasome is suggested by the demonstration of increased plasma concentrations of circulating IL-1β and IL-1826.
There is robust evidence indicating that HSP70 activates the inflammasome. Asea et al.34 demonstrated that HSP70 binds with high affinity to the CD14 receptor on human monocytes, inducing upregulation of TNF-alpha, IL-1β, and IL-6. More recently, Ullah et al.40 showed that knocking down HSP70 suppressed NLPR3 inflammasome expression.
Uric acid is a recognized DAMP signal that activates NLRP3 inflammasome with increased expression of caspase-1, IL-1β, and IL-1841-45. Indeed, inhibition of uric acid synthesis with xanthine oxidase inhibitors has been shown to suppress activation of NLRP3 inflammasome in several experimental models of kidney damage44. It has also been shown that uric acid, by increasing intracellular oxidative stress, drives the synthesis of HSP7046,47. Correlation between uric acid and HSP70 levels in hemodialysis patients has been reported recently48. Figure 1 summarizes putative mechanisms by which uric acid and HSP70 could interact in the pathogenesis of HTN.
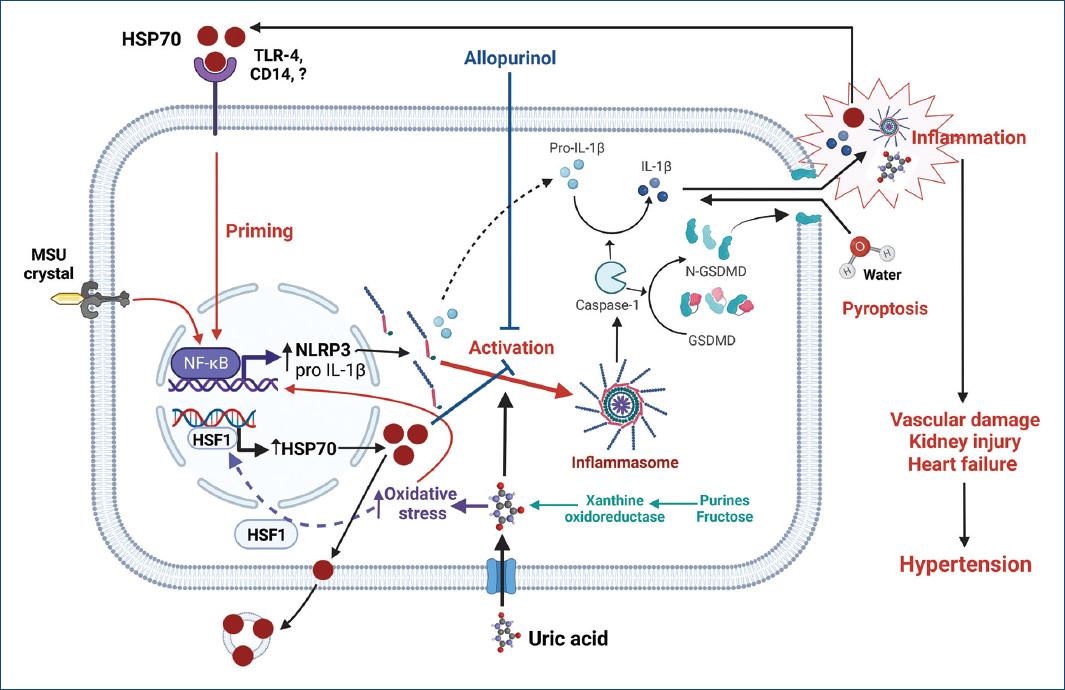
Figure 1 The increment of intracellular uric acid, either by over-production or uptake, increases oxidative stress and stimulates the generation of HSP70 to protect protein homeostasis. At first, intracellular HSP70 downregulates inflammasome activation; however, uncompensated oxidative stress induced by increased intracellular uric acid further drives the synthesis of HSP70. The excess of HSP70 is then exported to extracellular space through exosomes where it can bind various types of receptors (TLR4, TLR2, and CD14) and prime inflammasome activation. Monosodium urate crystals (MSU) and intracellular uric acid can drive inflammasome assembly leading to the activation of caspase-1 that mediates the generation of the active forms of IL-1β and other cytokines and the proteolysis of gasdermin. N-gasdermin fragments create membrane pores, cell swelling, and osmotic lysis (pyroptosis) with the extrusion of pro-inflammatory cytokines, damage-associated molecular patterns (DAMPs), such as uric acid, as well as other mediators that further increase inflammation. The synergic participation of uric acid and HSP70 favors the spread of inflammation that results in sodium retention by the kidney, overactivity of the sympathetic nervous system, impaired vasodilation in peripheral arteries, heart overload, and hypertension. Created with BioRender.com.CD14: cluster of differentiation 14; GSDMD: gasdermin D; N-GSDMD: N-terminal GSDMD fragments; HSF1: heat shock factor 1; IL-1b: interleukin 1 beta; MSU: monosodium urate; NF-kB: nuclear factor kapa B; NLRP3: NOD-, LRR-, and pyrin domain-containing protein 3; TLR-4: toll like receptor 4.
Conclusions
Inflammation is a central characteristic in the pathogenesis of primary hypertension. Hyperuricemia, HSP70 immune reactivity, and NLR3 inflammasome activation are common features in experimental and clinical hypertension. Potential interaction between uric acid and HSP70 may represent an important synergy in the development and maintenance inflammation.

