











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El tumor de Frantz fue mencionado por primera vez en el año de 1959 por Virginia Kneeland Frantz.1-3 El tumor sólido pseudopapilar de páncreas (SPT) es una de las neoplasias menos frecuentes, que representa de 0.2 a 2% de todos los tumores pancreáticos y de 1-2% de los tumores exocrinos. El tumor recibió varios nombres por sus características macroscópicas y microscópicas, hasta que fue definido como “tumor pseudopapilar sólido del páncreas” por la Organización Mundial de la Salud (OMS) como un tumor único, en 1996.1),(2,4,5
La mayoría de los pacientes con tumor pseudopapilar sólido de páncreas son del sexo femenino (relación femenino:masculino de 10:1), en la segunda o tercera década de la vida con un promedio de 22 años de edad; cerca de 20-25% se ven en edades pediátricas y sólo 6% de los casos se presenta en pacientes mayores de 50 años.1-4,6
Caso clínico 1
Paciente del sexo femenino de 16 años de edad, sin antecedentes heredofamiliares ni personales patológicos de relevancia para el padecimiento actual. Acude a consulta con cuadro clínico de dos meses de evolución, caracterizado por episodios intermitentes de dolor abdominal en epigastrio de intensidad variable, con irradiación hacia flanco y fosa lumbosacra del lado izquierdo, sin otra sintomatología agregada. A la exploración física con distención abdominal, dolor leve en epigastrio y sin masas palpables.
Se efectúa tomografía axial computarizada (TC) de abdomen superior simple y con doble contraste en cortes axiales, donde destaca páncreas con aumento severo de tamaño en cuerpo y cola secundaria a una gran lesión de bordes regulares, con importante efecto de masa sobre estructuras retroperitoneales, isodensa al parénquima, con áreas hipodensas no se refuerza al contraste intravenoso, sin calcificaciones ni áreas quísticas, de 102 × 107 × 115 mm (Figura 1), cabeza y proceso uncinado sin alteraciones, el resto del estudio sin alteraciones.
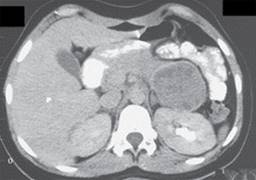
Los estudios de laboratorio (biometría hemática, pruebas de función hepática, amilasa, lipasa, tiempo de protrombina (Tp), tiempo parcial de tromboplastina (TpT), índice internacional normalizado (INR), electrólitos séricos, examen general de orina) estuvieron dentro de los parámetros normales. Se inicia protocolo de abordaje con sospecha de neuroblastoma como primera posibilidad diagnóstica, se solicitan catecolaminas totales y fraccionadas en orina de 24 horas, que resultaron dentro de los parámetros normales.
Se programa para laparotomía y se reportan los siguientes hallazgos: abordaje por incisión de Chevron, se abre la cavidad peritoneal y se realiza exploración protocolaria, se coloca separador automático tipo Balfour y se procede a abrir el espacio gastrocólico con el fin de entrar a la transcavidad de los epiplones. Se identifica tumoración dependiente de la cola del páncreas, al realizar resección de la misma para completar la extirpación del tumor y ligar los vasos de neoformación con bisturí armónico. Se procede a aplicar sutura corrida de poliglecaprone 25 de 2-0 en el segmento distal del páncreas, sangrado total de 200 cm3 aproximadamente, con un tiempo quirúrgico de 100 minutos.
Estancia hospitalaria de tres días con evolución favorable, toleró vía oral, con estudios de laboratorio completos donde únicamente destaca anemia grado 2 normocítica normocrómica. Egresa cuatro días después gracias a tratamiento antibiótico con ceftriaxona de 1 g IV cada 12 horas por siete días, y manejo analgésico con parecoxib de 40 mg IV, paracetamol de 1 g IV cada ocho horas y tramadol de 50 mg IV por cinco días, con cita por externo con resultado de estudio de patología.
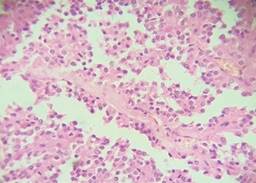
Estudio de patología reporta, al examen macroscópico, tumor de 13 × 11.5 × 8 cm sólido (Figura 2); y, al examen microscópico, presencia de pseudopapilas cubiertas por varias capas de células epiteliales (Figura 3). Cápsula íntegra, no involucrada por neoplasia. Se da seguimiento mensual con controles laboratoriales y de imagen, sin alteraciones.
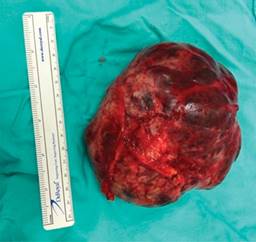
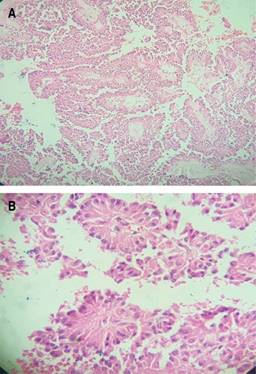
Caso clínico 2
Paciente femenino de 25 años, sin antecedentes heredofamiliares ni personales patológicos de importancia, que consulta por historia de tres meses de evolución con plenitud postprandial, vómitos de contenido gástrico sin pérdida significativa de peso, dolor abdominal referido en epigastrio e hipocondrio izquierdo tipo urente de leve a moderada, intensidad sin irradiaciones que se exacerbaba a la ingesta de cualquier alimento.
A la exploración física se aprecia leve distensión abdominal, peristalsis conservada con dolor a la palpación media y profunda en epigastrio e hipocondrio izquierdo con masa palpable en misma área, sin adenomegalias ni datos de irritación peritoneal.
Se realizaron estudios paraclínicos laboratoriales y de imagen, se encontraron laboratorios sanguíneos (biometría hemática, química sanguínea, pruebas de función hepática, Tp, TpT, electrólitos séricos, amilasa, lipasa) sin alteraciones, al igual que la radiografía de tórax; sin embargo, se solicitó TC de abdomen superior, inferior y pélvico simple y con contraste bifásico no contrastado con enfoque diagnóstico, y como único hallazgo se reportó “bazo de tamaño normal; sin embargo, existe masa con 40 unidades Hounsfield con calcificaciones en la pared, bien delimitada, localizada en el hilio esplénico que mide aproximadamente 8.2 cm por 6.6 cm” (Figura 4). Se decide intervenir quirúrgicamente.

Con abordaje tipo Chevron, se abre cavidad abdominal, que inicia con protocolo de laparotomía exploratoria enfocada al espacio gastrocólico y se ingresa a la transcavidad de los epiplones, donde se encuentra tumor de la cola del páncreas que se extendía al hilio esplénico y que respetaba retroperitoneo, el cual pudo ser resecado con la cola del páncreas junto con el bazo, al hacer tracción del estómago para exponer ligamento gastroesplénico a fin de encontrar de forma directa la transcavidad de los epiplones. Se cortaron y ligaron vasos en el ligamento gastroesplénico para darnos buena visualización de la arteria esplénica. Se cortó peritoneo sobre el bazo para facilitar ligadura de arteria y vena esplénica con hilo poliglactina 910 2-0, sin dejar algún tipo de drenaje. Sangrado 300 cm3 aproximadamente, no hubo necesidad de hemotransfusiones, tiempo quirúrgico 120 min.
En la evolución del postoperatorio el paciente tuvo tendencia a mejorar y después de cuatro días de ayuno, por elevación de amilasa secundaria a manipulación y sutura de páncreas, presentó tolerancia a la vía oral, se dio de alta con niveles normales de amilasa, además se le agendó cita después de un mes para continuar con la vigilancia clínica y conocer sus resultados histopatológicos.
El informe histopatológico diagnóstico fue concluyente y reportó una pieza de 9.1 × 7.6 × 7.2 cm, de superficie externa lisa, gris con red vascular visible y aumento en la consistencia con aspecto quístico (Figura 5); imagen histológica de tumor sólido pseudopapilar de páncreas (tumor quístico papilar-tumor de Frantz) (Figura 6), localizado en cola de páncreas sin observarse actividad tumoral en cara externa de cápsula de la lesión neoplásica, bazo con congestión sinusoidal.
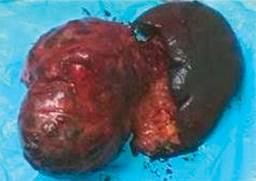
Discusión
El tumor de Frantz es un tumor enigmático, en cuanto a su origen celular y genotipo. Su predominancia en pacientes jóvenes del sexo femenino nos indica la posibilidad de una influencia hormonal en su desarrollo, sólo se han demostrado receptores de progesterona en estos tumores y algunos reportes sugieren la presencia de formas beta de receptores de estrógenos. Se han reportado pacientes infectados con virus de la hepatitis B, el cual puede inducir una sobreexpresión de β-catenina en las células tumorales, lo cual nos plantea una participación de este virus en la patogénesis de este tumor.4,7,8
El tumor pseudopapilar sólido de páncreas se caracteriza, desde el punto de vista genético, por la activación de β-catenina y sus células blancas. En 95% de los casos se muestran alteraciones de las vías del gen de la poliposis adenomatosa del colon (APC) β-catenina y de la ciclina-D1, con mutaciones activadoras en el exón 3 del gen de la β-catenina, lo que conlleva la acumulación nuclear y tinción positiva para β-catenina, en la mayoría de estos tumores. A diferencia del adenocarcinoma ductal del páncreas, el tumor de Frantz no se asocia con alteraciones en los genes K-ras, p53 ni DPC4.4
La mayoría de los diagnósticos de tumor de Frantz se realiza de manera incidental como un hallazgo dentro de estudios imagenológicos realizados por otras razones. Estos tumores causan escasa sintomatología hasta que alcanzan dimensiones importantes; presentan dolor abdominal en algunas ocasiones, distensión, saciedad precoz, anorexia, náuseas, pérdida de peso, pancreatitis e ictericia. Se han reportado casos raros de hemorragia intraabdominal por ruptura del tumor. El principal sitio de metástasis ocurre en el hígado y el bazo.3,4,9 Los tumores extrapancreáticos son raros y a veces no se demuestra tejido pancreático ectópico.4
La tomografía computarizada es el estudio de elección para la detección de los tumores pancreáticos, en el tumor de Frantz sus características tomográficas más relevantes son una ubicación aislada frecuentemente en la cabeza del páncreas, una ubicación mixta más frecuente en el cuerpo y la cola del páncreas, contenido predominantemente sólido, mayormente sin calcificaciones, tamaño predominante de 5-10 cm y de forma principalmente redondeada con bordes definidos.10,11
Desde el punto de vista histológico son lesiones encapsuladas con áreas sólidas y quísticas. La apariencia pseudopapilar se encuentra alrededor de un tallo fibrovascular. Las células tumorales poligonales forman áreas sólidas o se agrupan en pseudorrosetas. El estroma puede ser mixoide o hialino, pero con frecuencia es imperceptible. Los macrófagos espumosos son ácido peryódico de Schiff (PAS) positivos.7,10,11 Estos tumores se consideran de bajo grado de malignidad. Se recomienda la resección del tumor en todos los pacientes.1,2,4,7,9,12 Debe realizarse una resección oncológica con bordes quirúrgicos negativos para lograr el control local de la enfermedad, prevenir la recurrencia y las metástasis, aliviar los síntomas y asegurar un buen pronóstico a largo plazo.4
Ochenta y cinco por ciento de los pacientes presentan enfermedad local al momento del diagnóstico, 15% presentan enfermedad diseminada. Cuando la resección es completa, el pronóstico a largo plazo es excelente, con una supervivencia de 95% a los cinco años. Se recomienda seguimiento con imágenes postoperatorias cada seis meses durante dos años y después anualmente de por vida.4,10
En relación con lo revisado y en la experiencia de los casos reportados se mantienen resultados similares, al encontrar sólo síntomas gastrointestinales relacionados con la clínica. Como principal auxiliar diagnóstico sigue la tendencia de la TAC como método de diagnóstico; sin embargo, el hallazgo incidental transoperatorio es una variable que, en nuestra experiencia, se debe considerar.
El tratamiento quirúrgico es el de elección para una resolución completa de la patología.
Conclusión
El tumor de Frantz es una neoplasia infrecuente, usualmente de diagnóstico incidental y de bajo grado de malignidad. Se encontró mayor incidencia en el sexo femenino, en un rango de entre 15 y 30 años, y sin antecedentes identificados. Clínica y laboratorialmente es de muy difícil diagnóstico, lo que deja a la TAC como el principal método de imagen de elección para la identificación de estos tumores pancreáticos.
El tratamiento definitivo es la resección quirúrgica completa del tumor pancreático, lo que brinda un excelente pronóstico a largo plazo, aún sin reportar complicaciones relacionadas al tratamiento.

